食品開発展2025 |CTOプレゼンテーション公開&出展ブースにて頂いたQ&Aまとめ
はじめに
食品事業部の村瀬です。
株式会社digzyme(以下、弊社)は2025年10月15日(水)〜17日(金)に開催された「食品開発展2025」に出展いたしました。会期中は、「出展者・特別プレゼンテーション」に弊社のCTO、中村が登壇し、『差別化できる味・香り・食感の開発に向けたdigzymeにおける産業用酵素の研究開発事例』のタイトルにて、発表を行いました。
その内容を再録し、YOUTUBEで公開いたしましたので、お知らせいたします。
今回は、この度の展示会出展と動画の公開を機に、会期中にいただいたご質問の中から特に多かった内容をピックアップし、回答と合わせてQ&A形式でご紹介いたします。
「出展者・特別プレゼンテーション」の内容を受けて、さらに踏み込んだご質問をくださったお客様や、その他にもdigzymeの産業用酵素や開発プロセスに関してご関心をお寄せいただいた皆様へ向けた内容になっておりますので、宜しければぜひ最後までご覧ください。
Q1.
digzymeがin silico技術で設計した酵素を活用することで、これまで解決が難しかった香りや食感の開発課題に対して、どのような新しいアプローチが可能になりますか?
A1.
弊社では、既存酵素の特性をベースに活性や基質選択性などを向上・調整した新しい酵素を探索・デザインします。そのため、従来では難しかった味・香り成分の比率や、粘りや硬さなどの食感をより細かく制御することが可能になると期待しています。
Q2.
食品素材の味や香り、食感の改良以外には、酵素の働きによってどんな改変ができますか?
A2.
一例として、酵素による食品素材の物性改変が可能です。例えばパンやご飯のデンプンの構造を変化させることにより、時間が経って固くなるのを防いだり、植物性タンパク質を部分分解することにより溶解性を向上させることができます。
既存の酵素では、特定の素材や条件下で十分な改質効果を得られない場合もありますが、弊社ではご希望に合わせて目的の基質や環境条件に適した酵素を設計できるため、これまで難しかった素材に対する物性改変にもアプローチ可能です。
その他にも、酵素を用いることで原料素材から機能性の知られる食品素材に変換・合成することも検討しています。
Q3.
今後、in silicoでの酵素設計で、食品開発の持続可能性や生産効率を向上できる可能性はありますか?
A3.
in silico技術で最適化した酵素は、副生成物を減らしたり、より低温・短時間で反応する設計も可能です。その結果、エネルギーや原料の使用量を抑えつつ、安定した香り・味・食感を提供できるため、持続可能性や効率性の面でも大きなメリットが期待できます。さらに、従来は廃棄されていた副産物や原料も、適切な酵素処理により新たな製品に変換することができるため、食品のアップサイクルにも貢献します。
Q4.
digzyme Moonlightで設計した酵素を効率よく生産するための生産株開発は、今後どのような方向で進める予定ですか?また、将来的にはどんなことが可能になるのでしょうか?
A4.
今回のプレゼンテーションでは詳しく触れませんでしたが、酵素を実用レベルで安定生産するために、生産株の開発を進めています。具体的には、酵素を高効率に発現できる微生物株の選定や、酵素を大量生産するための培養条件の改良などです。将来的には、イノベーティブな酵素を、より高速に事業化していくためのプラットフォームとなる生産株構築システムを確立し、ニーズ対応酵素のデザインから事業化まで一気通貫でお応えできるようになることを目指しています。
終わりに
今回の食品開発展2025では、多くのお客様から具体的かつ本質的なご質問をいただき、酵素デザインへの関心の高まりを改めて実感いたしました。
弊社はこれからも、酵素が持つ可能性を産業界の皆様と共に開拓していきたいと考えています。今回のQ&Aがお役に立ち、皆様の新たな製品開発等のヒントとなれば幸いです。
活用されずに眠る配列データの宝の山を探索:鈴木彦有が語る、価値を引き出す解析の現場(社員インタビュー)
はじめに
本記事は、弊社のnoteに2024年11月に掲載されたインタビュー記事を、より多くの方にご覧いただけるよう、当テックブログに転載したものです。内容は掲載当時のものとなります。
(※記事中の組織名・役職等はすべて取材時のものです。※文中では、社内での呼称に合わせ「彦有さん」と表記しています。)
本文
「インフォマティクス×酵素開発」
東京工業大学で分子進化を学び、ビッグデータ解析の専門家としてdigzymeに参加した鈴木彦有さん。
今回は、研究開発の裏側から見た「データ駆動の酵素探索」の現場についてお話を伺いました。

ーー早速なのですが、digzymeご入社の経緯を教えてください。
はい。具体的に転職を考え始める以前 からJREC-IN(科学技術振興機構が提供するキャリア支援ポータルサイト)に登録していたのですが、そこにdigzymeの求人が出ていたのをたまたま目にして。
母校の東工大発だし、縁がありそうだなと思ったのと、自分自身のスキルの評価として、僕が持っているノウハウが使えて即戦力になりそうだなと感じたことが応募のキッカケです。
ーーなるほど。前職ではどんなことをなされていたのですか?
ゲノムとかトランスクリプトーム・・・いわゆるオミックス解析と呼ばれる、バイオロジーのビッグデータを対象とした、解析サービスを提供している会社にいました。digzymeと似てるところは、オーダーメイドに近い案件が多いところでしたね。
ーーdigzymeでは共同研究として受けているような案件に近いでしょうか?
近いです。もちろん酵素を対象にした開発っていうわけではなかったんですけれど。
ビッグデータを使った解析って、計算機のノウハウが必要なので誰にでもできるわけでは無いので、食品関係や製薬関係の企業さんなどから頂いた案件に対してそれぞれに応じた解析を行い、なんらかの結論を返す、というお仕事をしていました。コンサル業のような形をイメージしていただくとわかりやすいかもしれません。
ーー研究開発のコンサルティングに、バイオのビッグデータを絡めていた?
そうですね。そういうベンチャー企業に5年くらいいました。扱う案件としてわかりやすい例だと、やっぱり抗体医薬品関係。近年の製薬会社の売上高上位を占める製品は抗体医薬品ですから、関連した解析をすることが多かったです。
「抗体」は、特定の抗原(ターゲット)だけに結合する能力を持ったタンパク質です。
なので、抗体医薬品は従来の医薬品より副作用も減らせますし、がんや難病の治療時に強力な効果を発揮するものなのですが・・・抗体は「タンパク質」の一種なので 、もちろんベースになる「ヒトの抗体の遺伝子」があるわけです。
この抗体遺伝子をハムスターの培養細胞に組み込んで、大量に作らせて精製して、製品にする、という流れがあるんですが、その過程で、培養方法をどう工夫すればより効率的に大量に作れるか・・・
あるいは、新しい抗体医薬品を作るとしたら、どういう配列のものを作ればいいか、などを考える必要があります。これらを考察する時に、ビッグデータから知見を得るということが最近ではよくあるんですね。
例えば、少量のスケールならできていたことを、実生産の大スケールで行おうとしても、少量の時の単なる比例にはならない。100倍のスケールにしたら100倍できるかっていうとそうはいかないので(笑)そういう時に何が起きているのかを分析したりするために、生き物のビッグデータを取得して調べるアプローチがあり、それらのお手伝いしていました。
ーーなるほど。量産時の生物のコントロールは難しいと、中村さんもおっしゃっていました。
digzymeに入社後はどのような業務に関わってきましたか?
入社直後は、合成生物学の発想に基づく開発がメインでして、目的化合物を生き物に合成させるための、新規の反応を含む反応経路の探索プログラムの開発をしていました。
例えば出発化合物Aから目的化合物Dを作りたい、というときにAからBを経て、Cを経て、Dを作らなければならないとします。途中に何段階かの反応を噛ませないといけないということは、この間を繋ぐ反応経路の推定をしなければ、それらの反応を触媒する酵素探索ができないわけです。
ちなみに合成生物学では培養細胞や微生物を使って、何か人間にとって有用な物質を作りたいとなった時に往々にして一反応では済まないので、反応経路の推定は不可欠なんですよね。
ということで、色々な経路がありえるなかで、最適なものを探索できるようにしていました。
最近のdigzymeは産業用酵素をメインに扱っているので、お客様の求める酵素そのものを作れば良いわけですから、反応経路を気にする必要はなくなっています。
こういう反応を触媒する酵素が欲しいです、という酵素を提供できれば良いので、酵素のバリエーションをライブラリとして持っておくための開発に注力していますね。
ーーライブラリの開発にも触れていただきありがとうございます!最近のお仕事も教えてください。
DRY解析に詳しくないWETのメンバーにも扱える解析環境の開発・保守をしています。
ーー具体的にはどのような内容になるのでしょうか?
一般的にIDE(Integrated Development Environment/統合開発環境)の導入とい言われるところですね。いざ解析を始めようとすると、初心者にとっては解析ソフトウェアのインストールだけでも高いハードルになります。皆さんなんとなく『インストーラーをダウンロードしてきてダブルクリックすると、コンピュータに入る』みたいなイメージをお持ちかと思うんですが、バイオインフォマティクスでよく使われる多くのツールって、そうじゃないんですよ。インストールの段階から全部コマンドラインで文字列を打って、コンピューターに命令を出していくので、初心者がいきなりやろうとすると、まずこのインストールでつまづいてしまうくらいコンピューターに関する専門知識がある程度必要だったりするんですね。なので、そのような工程が不要になるように、なるべく環境ごと用意して渡せるようにしていく・・・というのが別に弊社でなくても、よくある解決手段なのですが、IDEの導入というのはまさにそれのことです。
さらに、バイオインフォマティクスの多くのツールはGUI(Graphical User Interface)が提供されていないのでより親しみにくいのですが、直感的に操作できるところまで持っていくのは大変にしても、jupyter notebookを利用することでこれもできるだけ解消するようにしています。
これはどういうことかというと、WETの方も後からのトレース用に『実験ノート』を作りますが、同じように『解析のノートブック』を作るんです。もちろんただのメモじゃなくて、実行コマンドが書いてあるし、押すとそのコマンドが実行されるボタンもついている。このような機能を整備することで、同じようなコマンドラインを別の案件で流したい時に役立ちます。
極端な話、一個一個の処理にどういうプログラムが使われているのか判っていなくても解析ができちゃうんですよね。もちろん全くわかっていないで触るのは問題なので、あくまで極論を言えばですが(笑)
田村さん(※注1:インフォマティクススペシャリスト、田村康一さん)や礒崎さん(注2:プリンシパルインベスティゲータ、礒崎達大さん)とも適宜協力してこのような環境を作っております。
ーーなるほど。ちなみにWETの方々はどのようなシチュエーションでDRY解析をされるのでしょうか?
まず想定されるのは、お客様とNDAを締結した後で、蓋然性を検討するフェーズですね。
理想的には、WETの研究員とお客様が面談している最中にささっと解析をして蓋然性の検討ができると、DRYの研究員メンバーがその場にいなくても簡単なご返答ができてとてもスムーズですから。
また、実際の事業案件として研究開発を進めていくフェーズでも、DRY解析の結果に基づいて実験対象や方針決めをWETの研究員だけで完結できることは開発速度の面でメリットが大きいと思います。
ーー教えていただきありがとうございます。その他にはどのようなお仕事をなされていますか?
個別の案件で実際の解析業務や調査業務を担当したり、新たな解析アルゴリズムを考案することもあります。最新の論文を読んで、社内の既存のツールの精度を高めるための実装に使ったり、バイオロジカルな方面で、判定のための指標値の計算について考えたりということがありますね。
ーーどんな論文を読むことが多いですか?
もともと分子生物学、分子進化学が専門なので、タンパク質のアミノ酸配列などを調べていったり、ゲノムとか、トランススクリプトームとか言われる配列と紐づく「量」の情報を扱うような解析が得意なので、遺伝子配列を直接扱うような論文を読むことが多いです。
先ほど説明した『ノートブック』のなかで、配列データの処理としてうまく使えるところがないかなどを検討します。
ーー同じインフォマティクス・スペシャリストの田村さんは構造的な側面の専門家ですが、彦有さんは遺伝子配列の側面から日々解析を進めてくださっているんですよね。
そうですね。ちょうど得意分野が分かれているので、digzymeでは酵素を多角的な視点から捉えた解析が可能となっていて、そこが強みの一つかなと思います。
ーーでは次に、digzymeで働いていて、どんなところにやりがいを感じられるか教えてください!
やはり、開発した成果が直接、お客様にとっての新たな価値や当社にとっての利益を生み出すための手段として使われる可能性が高いところですね。
『世の中のニーズに応えられるかのテスト』という側面からのPoC後も、ずっと長くお付き合いくださっているお客様も多いです。秘密保持上多くは語れませんが、digzymeとのシナジー効果が大きいと感じていただけているのかな、と。
今後もニーズに対して新しい価値を提供して、貢献していきたい気持ちがあります。
ーー彦有さんは、digzymeだからこそできる新しい価値の提供についてどのようにお考えですか?
冒頭でお話した内容とも重複しますが、まず前提として、酵素に関わる様々な遺伝子の情報がビッグデータとして既に存在しています。その中には、配列は取られているけど機能がよくわかっていないものが大量にある。
配列を取っている=天然に存在している=判っていないだけで、何かしらの機能を持っている、ということで、人類にとって有用な反応を触媒できるような酵素がまだまだ沢山存在する可能性があるということ。それをどうやって探し当てるか?というときに、一個一個実験をして探していくのは到底無理ですので。最初に『この辺にありそうです』という絞り込む作業を計算機上で行い・・・もちろん最後は実験をしなければいけませんが、なるべく少ない実験のなかで当てられる確率が高くなるようにしていく。ですから最初に絞り込む作業ってすごく重要ですし、digzymeの技術の本質はそこにあります。
我々が掲げる『世界を変える酵素を、迎えにいこう。』のミッション通り、(注3:2024年11月時点。現在は「ものづくりに、まだ見ぬ選択肢を。人と地球に、持続する活力を。」に変わりました。)これからもビッグデータのなかにある宝の山を発掘して、迎えに行きたいですね。
ーーdigzymeの技術の本質、掲げるミッションに関しても触れていただきありがとうございます!
ちなみになのですが、配列は取られているけど機能がよくわからないものが、ビッグデータ上に大量に登録されている背景に関しても教えていただけますか?
もともとは個々の研究者たちが研究している遺伝子の配列を登録することに始まり、それをあとで第三者が検証したり、検証とは別に、他の研究に再利用して使えるようにしていたんですね。ところが、DNAの塩基配列を解読するシーケンスの技術が飛躍的に向上したため事情が一気に変わってしまいました。特に2010年代からは次世代シーケンサーと呼ばれる解読装置が普及 してきてしまったため、それまでとはうって変わって、安価に大量の塩基配列を解読できるようになりました。なので、今までは目的の遺伝子だけを狙ってPCR法などで増やしてから塩基配列を読むのが普通だったんですけれど、そうやって特定の遺伝子を増やして読むのではなくて、『ゲノム全体を読んじゃいましょう!』とか『転写されているRNAを全て読みましょう!』といった感じで『とりあえず全部読む!』ということができちゃう時代に突入してしまったわけです。
そうすると大量に読みまくったデータが、どんどん溜まり続けていくんですよね、いろんな生き物で。
これらに関して、もちろん別の研究に流用できる期待値が高いデータだという側面はあるものの、データを取った人たちは結局、自分の研究に関わる部分しか見ない、というか見れないことが多いです。
一方で、長年研究をしていると、別の研究で取られたあるデータについて、自分の研究にとって有用かどうかという視点で見てると『使える』ということはよくあります。
なので、digzymeも研究者ならではの視点でデータの再利用性に着目するという発想に辿り着きました。これからも、まだ開拓されていないニーズに対して、将来的に応えられる可能性があるデータを埋もれているところから掘り起こす企業活動をしていきます。
ーーなるほど。背景についてもよく理解できました。様々な知見をお持ちの彦有さんですが、大学ではどのようなことを専門になされていたのでしょうか?ぜひ改めてお聞かせください。
学生時代は、東京工業大学(現・東京科学大学)の岡田典弘先生(東京工業大学名誉教授)と二階堂雅人先生(東京科学大学准教授)のもとで、進化についての研究を行っていました。
具体的には生物の遺伝子やタンパク質といった分子レベルのデータを使って、異なる生物間で遺伝子の進化的な系統関係を調べたり、そこから発展して遺伝子がどのように進化するか、また祖先の生物が持っていた遺伝子の機能を考察したりしていました。
僕が配属された当時、岡田研究室では東アフリカの三大湖(ビクトリア湖、マラウィ湖、タンガニイカ湖)に生息するカワスズメ科の淡水魚(シクリッド)に着目していたのですが、この魚は形態的・生態的に多様性に富んでおり、それぞれの湖に固有の種が数多く生息していることから生物進化や種分化(生物の種が分かれていくこと)を考える上でのモデルとして世界的にも研究対象になっていました。シクリッドが種分化を繰り返しながら莫大な多様性を獲得するに至った過程について、遺伝子から説明しようというチャレンジな研究が、研究室の内外で精力的に進められていたんですね。
この頃、同じく岡田研にいらっしゃった寺井洋平先生(総合研究大学院大学准教授)らのチームが行ったシクリッドの視覚関連遺伝子に関する研究などから、感覚に関係した遺伝子が先に進化することで種分化が促進されるという感覚駆動(sensory drive)仮説が提唱されており、これを受けて視覚以外の他の感覚についても感覚駆動仮説を検証しようと、二階堂先生のチームでは嗅覚に着目した研究を進めていました。
僕はそのなかでも特に、外界にある様々な匂いの物質をキャッチしてその刺激を細胞に伝えている嗅覚受容体遺伝子に着目し、その進化について調べていましたね。
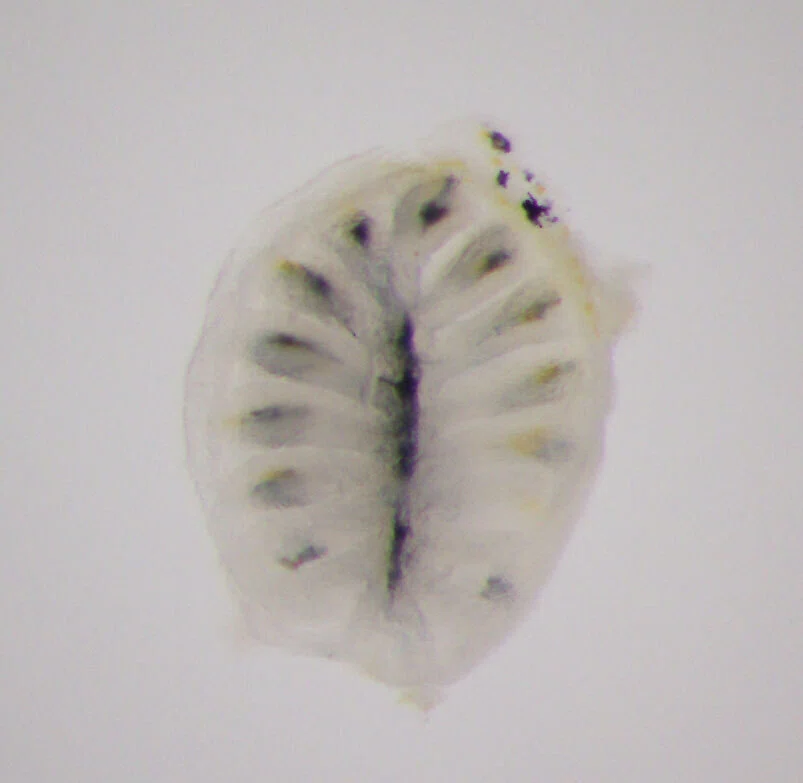
魚の鼻の穴を切り開くと、嗅房(olfactory rosette)と呼ばれるひだが沢山ある花びらのような構造体を肉眼でも見ることができるのですが、実はそこに匂いを感じる嗅神経細胞が多数集まっていて、魚が水の中の匂いを感じるための器官となっています。
これらの嗅神経細胞は一つ一つが別々の嗅覚受容体遺伝子を発現していると考えられているのですが、多数の嗅覚受容体遺伝子を分子系統学的に分類し、系統ごとに発現している嗅神経細胞を可視化したり、嗅覚受容体遺伝子以外にも嗅神経細胞に発現する嗅覚マーカータンパク質を使って、それらが発現している嗅神経細胞を可視化するといったWET実験に取り組んでいました。
そういったことに取り組むうちに、シクリッドに関する研究テーマからは少し離れて嗅覚受容体遺伝子や嗅覚関連遺伝子そのものの進化を調べていく方向に研究がシフトしていった感じです。
いくつか研究成果を紹介させていただきますと、ゲノムデータの解析から多くの脊椎動物は1つしか持たない嗅覚マーカータンパク質の遺伝子を真骨魚類は2つ持つことを二階堂先生が発見していたのですが、僕がその2つの遺伝子の発現を細かく調べたところ、それぞれ発現している嗅神経細胞が異なっており、さらに1つは嗅神経細胞以外に目の中の網膜の水平細胞にも発現していることが明らかになりました。
それまで嗅神経細胞のみに固有の発現を示すと思われていた遺伝子が、実は網膜の神経細胞でも使われていることがわかったのは面白いと思います。
また、シーラカンスのゲノムデータの解析に取り組んだ際、従来は陸上動物型と考えられていた嗅覚受容体遺伝子のいくつかの系統群について、実はシーラカンスやポリプテルスといったいわゆる「古代魚」と呼ばれる古いタイプの魚類にも数十個程度は存在することや、古代魚を含めた脊椎動物全般が保持している未知のⅠ型フェロモン受容体遺伝子(ancV1R)を発見したりもしました。
ancV1Rの発見については論文が出版された際、東工大のプレスリリースでも公表させて頂きまして、自分の中で特に思い入れのある研究です。

そんななか、あるときから次世代シーケンサー(数千億から1兆にまでのぼるDNAの塩基配列を短時間で解読する装置)から出力されるデータを用いた解析も行うようになりました。それにはいくつかきっかけがあったのですが。
もともと僕がいた岡田研はゲノムや個々の遺伝子の 配列を調べる解析を用いることが多く、バイオインフォマティクスを独学で学んで研究に使っている助教の先生やポスドクの先輩も何人かいらっしゃったので、じゃあ僕もちょっとやってみよう、と。
ーー独学で。
はい。コンピュータのプログラミングは、小さい頃に『BASIC』を遊びで触っていた経験もあったので、文法が似ているプログラミング言語のPerlを使い始めました。今ではあまり使われなくなってしまった言語ですが、当時はWebアプリケーション開発でもよく使われていて、文字列処理を得意とする言語ということもあり、塩基やアミノ酸の配列データ主に扱うバイオインフォマティクスにはうってつけでした。
そういうことをしているうちに、研究室では「バイオインフォマティクスができる学生」として認識されていきました。
その後、長谷部光泰先生(基礎生物学研究所教授)を代表とする「複合適応形質進化の遺伝子基盤解明」(2010年度から2015年度まで)という文部科学省の新学術領域研究公募テーマに岡田研が提案した課題が採択されまして、その際「バイオインフォマティクスができる学生」として岡田先生や二階堂先生の推薦もあって、ありがたいことに僕も参加させて頂けることになりました。
このプロジェクトは、当時普及し始めていた次世代シーケンサーを積極的に用いてゲノムやトランスクリプトームといったバイオのビッグデータを実際に取得して活用することで、あらゆる生物の複雑な進化現象の一端を解き明かそうというチャレンジングなテーマに挑戦するものでした。
それとともに、僕のような若手研究者に次世代シーケンサーのデータを扱うノウハウも含めたバイオインフォマティクスの技術を教授するといった教育的な側面もあったと思います。
実際、このプロジェクトに参加させて頂いたおかげで、これらの経験やスキルを積むことができましたし、それが前職そしてdigzymeの仕事でも活かされていると思います。
ーーなるほど。詳しく教えていただきありがとうございます。
WET,DRY両方に深く関わってこられた彦有さんですが、digzymeのなかでは今後どのようなことにチャレンジしていきたいですか?
まさに、WET &DRY二刀流の人材育成ですね。WET側がDRYを知ることで研究の速度が向上したり、DRY側がWETを知っていることで生命科学の現場に合った開発ができるようになると期待されます。
実は前職でも、バイオインフォマティクスの解析技術のハンズオンセミナーに講師として呼ばれて、企業やアカデミアの方に向けての技術解説をしていた経験があるので。
digzymeでも社内セミナーのようなものができたらいいなというイメージを持っています。
ーー社内セミナー、いいですね!
はい。IDEを作ったのも布石になっています。
もちろんIDEを使用するだけではあまりノウハウにはなりませんが、今後はWETの新人さんが入社したタイミングで、ファーストステップとしての『IDEを用いた解析をやってみよう!』というようなセミナーも開けたらな、と。このあたりはCTOの中村さんとも相談して進めていけたらなと考えています。
ーーDRYの技術者にバイオ出身者が多いdigzymeだからこそできる教育ですね。そういった意味でも人材の揃い方は希少価値が高いですよね。
そうなんです。
digzymeはむしろ、純粋な情報科学の人は採用していないんです。なぜならそれだと『酵素』の開発はできないから。
渡来さん、中村さん、田村さん、僕、みんなバイオに関係する分野をバックグラウンドとしており、バイオ側から情報科学の知識を仕入れていった経験があります。
WETの皆さんはもちろん、今バイオの領域にいらっしゃるわけですから、僕らのようにバイオ側から情報科学を取り込むという方向性に進むことが可能ですし、その育成ができるのがdigzymeだと思っています。
本来こういった人材育成は日本の国策としても必要なので、業界全体で解決すべき内容だと僕は考えています。ですが、現実的に、WET出身者がDRYの技術を学ぶのって結構大変なんですよね。
一番の理由は、必要に迫られる機会が少ないからだと感じており、それこそdigzymeのように『バイオと情報科学両方がわかっていないと、開発が進まない環境』にでもいない限りなかなか難しいはずです。
ーー環境に左右されやすい?
といった側面もあります。
むしろ実験中心の研究室だと、実験もせずにパソコンに向かってカタカタしているだけだと遊んでいると見なされるというような話も聞いたことがありますし。
でも実際は、情報科学の人があえてバイオを勉強しないのはわかるんですけど、バイオの人は本当は情報科学とは切り離せないんですよ。
次世代シーケンサーで大量に配列を読むことが当たり前の時代になったので、いつまでもそれを避けているというのは、バイオの研究においてディスアドバンテージでしかないんです。
DRY部分は共同研究先にお任せすればいいという考えもありますが、そうするとバイオインフォマティクスができる先生方がとても忙しくなってしまうので(笑)
アカデミアでも本当は、各研究室でその生物を研究している方が自らDRY解析もできた方がいいと思っているのですが、これについては先ほどもお話しした進学術領域研究でコアメンバーだった先生方も当時から同じような理想を掲げておられたと思いますし、CEOの渡来さんも同じ意見だと思います。
ーー最後に、digzymeに応募を考えている未来の仲間に一言あれば、お願いします。
自身の探究心と他者への貢献の意志を両方持ちながら、仕事のできる環境があります。
ーー彦有さん、ありがとうございました。
終わりに
▼オリジナル記事はこちら(note)
https://note.com/digzyme/n/n7fbee270334b
研究推進と社会実装をつなぐコーディネーション実践:礒崎が語る課題解決の現場(社員インタビュー)
はじめに
本記事は、弊社noteへ2024年10月に掲載されたインタビュー記事を、より多くの方にご覧いただけるよう、当社公式テックブログにも転載したものです。内容は掲載当時のものとなります。
(※記事中の組織名・役職等はすべて取材時のものです。)
本文
ーー第1回目のCTO,中村さんのインタビューに続きまして、今回はPI(プリンシパル インベスティゲーター)の礒崎さんへインタビューをさせて頂きます!礒崎さん、よろしくお願いいたします。
早速なのですが、digzymeご入社の経緯を教えていただきたいです!学生時代は、東工大から京都大学の博士課程に進まれたんですよね。その後、digzymeに入社されたとか。
はい。京都大学の博士課程を中退したのですが、その後東京に帰ったタイミングで、東工大で1つ上の先輩だった渡来さん(※digzyme代表取締役CEO、渡来直生)がdigzymeを発足させていて。『面白そう』だったので声をかけたことがきっかけです。
ーーそうだったんですね。東工大時代、お二人はどういうご関係だったのですか?
そもそも渡来さんと知り合ったのは、東工大時代のiGEM(※The International Genetically Engineered Machine Competition)で・・・
ーーiGEM!digzymeも協賛していますよね。
はい。iGEMは、まだ学部生でも研究室に入らず擬似的に研究活動がちゃんとできる、世界最大規模の合成生物学コンペティションです。
そのiGEMで、渡来さんとは東工大代表として同じチームだったんですね。3人1組のチームと少人数だったこともあり、当時から結構仲が良かったんです。
ーー3人1組だとかなりの時間を一緒に過ごすことになりそうですね。
ですね・・・というようなこともあり、渡来さんがすごいというのは当時から知っていたんで(笑)
ーー『すごい』ですか、、、なるほど(笑)
もう、iGEMの時からキレッキレだったんで(笑)時を経て、この人が会社をやるんだ・・・!これは面白いことになるぞ、と。
ーーあ、『「面白そう」だったので声をかけた』の面白さって、そこですか?(笑)
そう・・・あの渡来さんがCEOである・・・というところですね。
そもそも僕は、学生自体に専攻していた分野的に、当時は酵素についてそこまで詳しくなかったので。
東京に帰ってからしばらくは他の会社で技術営業みたいなことにも携わっていたのですが、少しの兼業期間を経て、その後digzymeにフルコミットする形になりました。
ーー他社様で、技術営業もされていたんですね!digzymeへフルコミットするに至った決め手は何だったのでしょうか?
技術営業のお仕事では、取り扱っているプラットフォームの説明をお客様にして、いったん社内に持ち帰って、対応する技術員に渡す、という立ち位置だったんですね。
それ自体は大切なお仕事ですし、むしろdigzymeでも今、人材が欲しいポジションでもありますが。ただ、ほとんどお客さんとのネゴシエーションがメインで、新しい技術的なことに直接携わることが少なくて。僕としてはちょっと、橋渡し役的なところに無力感を感じてしまっていたんです。
技術営業ってこういうことなんだ、というのが体感的に理解できたのはとてもいい経験だったのですが。
研究者のクリエイティビティって、自分で課題を設定して勝手に自分でゴールに向かって走っていくところにあると思うので、digzymeではそれができると思ってフルコミットに至りましたし、実際に今、そのようにできているのが嬉しいです。日々の課題に取り組むことに、すごく達成感を感じております。
ーーなるほど。礒崎さんの望む方向に進まれていて、聞いていて私も嬉しく感じました!具体的には、どんなことに特にやりがいを感じますか?
共同研究や新規事業の課題を解決するための解析技術を日々作っておりますが、そのなかで生物学的な特徴をドライの解析にいかにうまく落とし込めるかということを話し合い、それを現場で試せるところですね。
ーー生物学的な特徴を、DRYの解析に落とし込むってどういうことなんでしょうか?
そうですね。具体的にいうと、お客様から頂く『抽象的な課題』をめちゃくちゃ細かく『生物学的にはこういう特徴のはず!』とDRYで検証していって、最終的にはWETに繋げていく・・・これを“落とし込んでいる”と表現しています。
手順でいうと、まずは、生物学的な特徴のどの側面が、解決したい課題に寄与しそうか?というところを、DRYの要件として落とし込んで解析していきます。
構造上的な形の差は?一時配列上のモチーフのパターンはどう違うか?生物種的に発現させることができるかどうか?などについて『仮説を出して、解析する』ということをDRYでは行いますが、それだけで終わらず、ラボでWET検証したり、共同研究の場合は先方様に実際に試して頂いたり。
共同研究、自社研究ともに仮説検証がフルセットでできる・・・つまり、頑張って考えた仮説を実証できるんですね。これが、僕にとっての大きなやりがいです。
ーー詳しくありがとうございます。
中村さん(※取締役CTO、中村 祐哉さん)のインタビュー時もでしたが、礒崎さんとお話ししていてもdigzymeはDRYとWETがシームレスに結びついていることをひしひしと感じますね。
そうですね。digzymeはDRYとWETという二つの基盤がしっかりあるので、非常に良い環境です。
ーーさて。礒崎さんは、DRY、WET、両方に精通していらっしゃいますが・・・日々の業務で、これまでのご経験が役に立っていると感じる場面はありますか?
それでいうと実は、DRYに関しての経験って、学生時代はユーザーとして、市販の解析ツールをちょっとだけ使える、くらいだったんですよね。コードを書いたりはできなくて・・・
ーーえ!信じられないです・・・!
そうだったんですよ。
しかも、どういう理屈でDRYのツールが作られているのかも知らないで、ほんとに『使えるだけ』って感じでした。なので当時は『DRYの人』とは呼べないですかね(笑)そもそもWETの人間でしたし。
博士課程では沖合に出て、海のバクテリアを調べてたりしていたんで。あくまでメインはWETで、補助的にLinuxをちょっと使う、というくらいでした。
ーー沖合・・・?海のバクテリア・・・?という気になる単語も出てきたんですが・・・。
ええと、当時はDRYのツールは、どんなことに使っていたんですか?
RNA-Seq(※次世代シーケンス(NGS)解析のひとつで、「トランスクリプトーム」と呼ばれる遺伝子発現を網羅的に検出する次世代シークエンサー)などを用いて、海のウイルスの感染対象をゲノム側の遺伝子から推定することなどに使っていました。
ーーなるほど。修士課程まではいかがでしたか?
修士まではLinuxも知らなかったですし、パソコンもwindowsだけで、エクセルくらいしか開いたことないような状態でした(笑)
というか、その頃はもっと遠くて、ゲノムすら扱っておらず。
修士ではマウスを使った癌のモデルなどの研究をしていたので、マウスに癌細胞を移植しては、そのサンプルをとって・・・職人技みたいな感じで、腫瘍の切片を作成して・・・顕微鏡で観察して・・・
みたいなことをやっていました。
イメージとしては、病理医の方が人間になさることにちょっと近いかもしれないです。
ーー今お仕事でなされていることとは、だいぶ違いますね。そして、確かに一切パソコンが登場していません・・・。
そうなんです。ただ、流石に学会などに足を運ぶなかで、DRYが全くできないと、アカデミアにせよ他の研究職に就くにせよ、生きていけないな〜と実感しまして。
ーーそういうものなんですか?
うーん、その感覚はWETを専門にやっている皆、ある程度持っている気がしますね。
「可能ならやっぱり勉強しておきたいな」
みたいなことは、今のdigzymeのメンバーも大半が口にすることだと思います。
ーーDRY、WET、両者を理解できているとメリットも色々ありそうです。
はい。でも何していいかよくわからなくてペンディング、みたいになりがちなんですが。
なので、僕も修士から博士に進む過程で、あえてラボを変えた意図はその辺にありました。
一応WETもDRYも両方できるであろうラボに移ったわけですね。とはいえ、先ほど申し上げましたように、市販のツールを使う程度だったので、digzymeに入社するまではコードを書くという意味ではpythonも触ったことがない状態でした。
なので、digyzmeに入ってから全部、渡来さん、中村さんに教わったんですよね。当時、某ファミレスで(笑)プログラミングを教わる会というのをたまに開いてもらって、宿題を出してもらい、ちょっとずつ上達する、みたいな。
ーーそうだったんですね。ちなみに、WETの経験があるからこそ、DRYの開発に活きてくる部分もあるんでしょうか。
そうですね。WETの人たちがDRYのツールを使うときの気持ちがわかる、という部分はかなり大きい気がします。せっかくなので、関連して、そもそもWETでやっていることをDRYでやる、ってどういうことなのか?ということも説明しましょうか。
まず、WETの「実験」部分をDRYで行うのは無論無理です。
DRYで行えるのは、それ以外のいわゆる調査の部分。例えば、digzymeだと『酵素の探索』がまさにそれなんですけれど、『今回のお題で、何の配列を出したらいいか?』っていうのは、本来は『研究職』という大きい枠の中の人の、作業のひとつです。
ですが、digzymeはDRYとWETという二つの基盤があるので、この『酵素の探索』はDRYの人が担当しています。どうやったら良い配列が取れそうか?という曖昧な内容も、DRYで自動化してサクッと判断できるようにしておくと、そのあとすぐWETで実験できますし、また、実験の数も減らせるというようなメリットが沢山あります。
『酵素の改変』も同じですね。『ここを改変したらいいよね』っていうのがDRYですぐわかる。でも本来は、これら全てが大きな枠での『研究職の人』の仕事なわけです。なので、WETの経験があれば自ずとDRYの開発にも活きてきます。
ーーなるほど!理解できました。ありがとうございます。
話は少し戻りますが、博士課程でなされていた、海のウイルスの研究についてお聞きしてもいいでしょうか?
海に関わらず、変なところにいるウイルスの研究がしたかったんです。そのなかで、先ほども申し上げました通りDRYもできそうな研究室を選んだら海洋分子微生物学研究室という選択になりました。
ーー『海の』ウイルスという部分にはそこまでこだわりがなかったということですね。
変なところにいるウイルスの研究がしたい!と思われたキッカケって何だったのでしょうか?
そうですね(笑)その理由を説明しようとすると、小学生の頃にまで遡るんですが・・・
当時ちょっとだけ話題になった『the FUTURE is WILD』(※人類滅亡後に、どんな生物が地球上で栄えるのかシミュレートした話題の書。象と見違えんばかりの巨大イカなど出てくる生物のCGも魅力的。/ドゥーガル・ディクソン (著), ジョン・アダムス (著), 松井 孝典 (著), 土屋 晶子 (著))という本がありまして。
そのなかで未来の生物が想定して描かれていたりして・・・また、逆に、古生物の本が好きだったり・・・生物の進化上、古生物を追っていくとどんどん小さくなるんですけど・・・最初、微生物からスタートして大きくなってく・・・その過程が、我々が生きている今の状況と全く違うので、単純にモンスターチックで面白いなと(笑)
なので『極限環境微生物』みたいな分野があるのでそれでもよかったんですけど、それよりもう一歩捻った感じのものが好きになってしまいまして。
例えば、ウイルスが哺乳類を作ったかもしれないという説があったりするんですが。
胎盤を作る部分の遺伝子が、レトロウイルスというウイルスで構成されていたりするんですけど、実はそれが急に生まれた原因はウイルス進化説に基づいている・・・とか。そのなかでも、特に変わったウイルスを扱うのは面白そうだなということが進路選択のキッカケになりましたね。
ーーウイルスって何者なのかは私も気になります。かなり奥深いんですね。さて、話題は変わるのですが、これまでに、仕事で苦労されたことがあれば教えてください。
お客様からの抽象的な課題を「事業的に有益、かつ、実現可能な解析手段」に落とし込むことですね。お客様にとって
『digzymeにどれくらいの粒度で依頼をしたらいいのかわからない』もしくは『そもそも粒度を細かくできない』というときに、課題が抽象的であることがしばしばあります。
例えば、『具体的な化合物の構造すらなくて、なんとなくこんな感じの性質を持ったものが欲しい』『なんとなく今作っているこの生産品の生産量が上がると嬉しい、酵素が使えるか使えないかわからないけど・・・』などのオーダー。
これらを実現する際、従来の手法や競合他社と、digzymeならではの技術を比較し、本質的に勝る部分があるかはもちろん、市場とマッチするかなども含めて多角的な視点で考え、最終的な解析手段に落とし込むことは毎回それなりに苦労しますね。
ーーなるほど。いつも、どのように乗り越えられていらっしゃいますか?
早めに社内外の適切な人材に相談することで解決しています。
また、今は事業部のメンバーが増えたので、僕が初回の面談から参加することも少なくなってきていて、上記のような抽象的なオーダーに僕自身が直面する機会は減っています。
会社の仕組みとして、抽象的なお題を具体化していくレイヤーがどんどん整ってきていていて有難いです。
ーーメンバーが増えることで、課題を解決しやすい環境がどんどん整っていっていますよね。
ところで『社内外の適切な人材に相談』といえば、礒崎さんは社内でかなり積極的にコミュニケーションを取られているイメージです。私もいつも助けられておりますが、社外でも?
そうですね。新規事業のお話でも、ビザスクなどで有識者の方にヒアリングをよくします。
特に食品事業部関連の内容だと、宮内さん(※取締役CSMO、宮内 琢夫さん)経由で有識者の方々を募って、ご意見を伺うことも。
ーー心強いですね。ヒアリングした内容で、心に残っているものはありますか?
沢山ありますが、一例として・・・
とある酵素(試験に使うような)の活性をすごくあげられるとしたら、それが事業になり得るかどうか?というお話をお聞きした際も、
「その酵素の活性は今まで一定以上上がったことがなくて、もちろん上がれば上がっただけ試験の精度が上がるので良いが・・・そんな例は技術上いままで見たことがない」
というお言葉をいただけました。
ーー「そんな例はみたことがない!」これは嬉しいですね!
はい。自信を持って、
「これは市場に深く刺さるなぁ」
ということを判断することができました。
ーー素敵な事例を教えていただきありがとうございます。
続いてのご質問なのですが、仕事をする上で大切にしていることはありますか?
これも、社内外問わずまめにコミュニケーションをとること、ですね。
そのためにも日頃から、問題が生じたときに早めの相談がしやすい関係性を築くことを大切にしています。
心理的障壁が下がっていると、悪い関係にはなりづらいですし『壁がある』と相手に明示しないことに気を配っていますね。
壁があることで、敵/味方、という2サイドが生まれてしまうので。ニュートラル以上であれば敵になることはないですし、問題が発生しづらくなるなと思っています。
ーー感慨深いです。そんな礒崎さんですが、今後どのようなことに取り組んでいきたい(チャレンジしたい)ですか?

まさに今取り組んでいることですが、wet研究業務全体の管理です。
過去はCTOの中村の下に社員がフラットに存在している、という状態でしたが、今年からは僕と高山さん(※PI=プリンシパルインベスティゲーター、高山 裕生さん)が全WETメンバーのひとつ上のPIというレイヤーとして入って、その下にPM(プロジェクトマネージャー)、DRYメンバーは中村直下という組織図になりました。
WETの皆の研究業務を直接管理することが今までなかったので、不慣れで少し苦労していますが、同じPIの高山さんにも助けてもらいつつ進めています。
今後のdigzymeに対して期待したいことは、さらにPM層が厚くなることです。なるべく、ある程度上流(DRY)のことがわかるWETメンバーを増やしていきたいですね。
ーー次回インタビュー予定の彦有さん(※インフォマティクススペシャリスト、鈴木彦有さん)も、先日少しお話しをした際に、同じことをおっしゃっていました。
とはいえ、いきなりDRYのことを学ぼうと思っても具体的に何を学んだらいいのかピンとこないと思うので、そこはOJTを通して伝えていけたらと思います。
もちろんWETメンバー全体に対してアプローチしていくよう心がけておりますが、早期に動き出しているという意味では、今は僕が根岸さん(※シニアアソシエイト、根岸 孝至さん)高山さんが崎濱さん(※シニアアソシエイト、崎濱由梨さん)にDRYのことを色々と伝授しております。
ーーなるほど。DRY解析ができるWETメンバーが順調に増えたら、さらに強い組織になりますね。他には何か挑戦したいことはありますか?
『三次元構造を活用した酵素ファミリー』の特徴を反映した解析パイプラインの構築を促していきたいです。
これによって、酵素業界全体でいままで埋まっていない需要や、これまで試されていなかった新規のアプローチに繋がるはずなので、酵素を基盤としたものづくり全般の多様化、生産性の向上が見込めます。ということで、田村さん(※インフォマティクススペシャリスト、田村康一さん)と一緒に進めていけたらなぁ、と。
digzymeの強みは、上流も下流もできて、さらに生産まで行えるように準備しているところにあります。『AIでちょっとした解析をしてくれるところ』というニュアンスとは根本的に違うので・・・
ーー強みを強みたらしめる動きになりそうですね。
はい。やっぱりdigzymeは、マーケットインが意識できているディープテックだというところが最大の特徴なのかなって。特に役員人はみんな、当然のようにスケーリングを意識しています。
ーーもちろん、細かい技術に関して他と差別化できているのは、
各分野のプロフェッショナルが集まっているからというところにありますが・・・ーー技術上、今できるかは一旦置いておいて、こういうことをすると大きくビジネスが飛躍していくでしょうというところにものすごくアンテナを貼っています。だからこそ、必要な技術が定義できて、定義した技術を確実に作れるという好循環があるんです。
ともすると、ディープテックの企業はスケーリングが目的ではない場合もあるんですよ。
でも僕らの考えは、『ある程度、稼げればいい』『この技術はすごいからどうせ引くてあまたでしょう』などのようなふわっとしたイメージじゃ駄目だということ。
いくら凄くても、使いたくない用途のものだったら『確かに革新的だけどな・・・』で止まってしまうこともよくあるそうですから。
ーーマーケットイン思考があるから、お客様の心を動かせるのかもしれませんね。
そうです。そのためにも渡来さんを筆頭に、スケーリングを意識してビジネスの最先端のところを調査し、それをちゃんと技術的な言葉に変換できる人がいることがすごく重要です。
ーー最後に、digzymeに応募を考えている未来の仲間に一言あれば、お願いします。
人間性も、研究やこれまでの業務のバックグラウンドも、多種多様な人材が揃っていて、研究だけにとどまらない様々な分野の経験をできるので、新しいことにどんどんチャレンジしたい方、歓迎です!
ーー礒崎さん、ありがとうございました。
終わりに
▼オリジナル記事はこちら(note)
https://note.com/digzyme/n/n4cb24197110b
探索・設計・発現をつなぐ基盤技術開発と人材育成の現場:CTO中村の視点より(社員インタビュー)
はじめに
本記事は、弊社noteへ2024年9月に掲載されたインタビュー記事を、より多くの方にご覧いただけるよう、当社公式テックブログにも転載したものです。内容は掲載当時のものとなります。
本文
ーー記念すべきインタビュー第1回目です。中村さん、よろしくお願いいたします。
早速ですが入社の経緯を教えてください。・・・といっても、中村さんは創業メンバーですよね。
そうですね。東京工業大学在学中に株式会社digzymeを、渡来さん(注1:渡来直生さん、digzyme代表取締役CEO)、山田先生(注2:東京工業大学准教授山田 拓司先生、digzymeの取締役CSO)と共同創業しました。
創業のキッカケとしましては、digzymeの創業の元となった研究を、山田研(注3:東京工業大学生命理工学院 山田研究室)で実施していて、ビジネスの可能性を感じたことです。
ーー創業の元となった研究・・・是非、もう少し詳しく教えてください。また、どんなところに
ビジネスの可能性を感じたのでしょうか?
現在のdigzyme Moonlight™の元にあたる研究で、『酵素を探索します』という内容でした。
長瀬産業さんと山田先生が共同で研究をしていたところに、僕も入って追加で解析をしました。
具体的には長瀬産業さんから
「こういう酵素が欲しい」
というオーダーをいただき、それをデータベースから探す・・・ということをやっていて。それで順調に酵素がみつかっていたので、まず論文にしましょうということになりました。
その後、もしこの探索技術を『他の部分』に適用したらどうなるのか?という考察を深めていったんです。
ーー他の部分とは?
この世界でまだ触媒する酵素が見つかっていない、化合物の合成反応をみつけられるのか?というところですね。これが長瀬産業さんとの研究で成果が上がりまして。ある植物が持っている『特殊な化合物を作るための酵素』についての研究だったのですが。毎回その酵素を、植物をすりつぶして抽出するのは大変じゃないですか。
なので、微生物の酵素反応を使って、酵素化学的にできたら簡単なのでは?ということで、それが可能な酵素を探索してみたんです。そしたら、結構たくさん見つかったんですね。
それも、まだ発見されていない、世の中では売られていないような化合物を合成するような酵素が、結構たくさん。
ーーすごいことですね。
はい。それからは、作るのがすごく難しい化合物を、これらの酵素を使えば、実は簡単に作れるんじゃないか?ということを論文のなかで議論して。
山田先生に報告したところ、こういった酵素が欲しい人々は世の中にかなりいらっしゃるんじゃないか?という話になり、ビジネスとしてやったら面白いのでは、と。
さらにタイミングがすごく良かったのですが、その当時、ちょうど渡来さんが、ベンチャー企業の立ち上げに興味をもって活動していて。
そのような流れもあり、山田先生と、渡来さんと、僕とで活動をスタートしたら早速、『そういう酵素を探す技術があったら使いたいです』というところから、複数お声がけ頂いたんです。なので、これだけニーズがあるならば創業した方がいいだろう、ということで創業に至りました。
ーー確かに、従来の酵素開発では、目的に合った酵素の遺伝子を見つけるまでに、膨大なトライアンドエラーが必要でした。
そのため、偶然の発見に頼らざるを得ない不確実性と、莫大な開発コストが問題となっていましたが・・・この点を考えると、かなりのニーズがありますよね。
創業後はどのような業務に携わってこられたのですか?
創業直後は、コアテクノロジーにあたる酵素探索ソフトウェアの開発作業を行いつつ、次はそれをさらに応用して、より会社の強みになる部分をどう開発していくか?というところを進めていきました。すでに研究開発のテーマもいくつか自社内であがっていたので、それらの開発を含めて。
でも僕は、創業から半年間はまだ学生だったのですが、卒業後は2年近く製薬会社に勤めていたんですね。なのでその期間は兼業しながら技術開発のディスカッションなどを中心に参加していました。
ーーその後、digzymeにフルコミットする形になられたんですよね。
はい。digzyme1本になったあたりで、ちょうどNEDO(注4:国立研究開発法人 新エネルギー・産業技術総合開発機構)のプロジェクトが始まりました。
そこでspotlight(注5:酵素機能改良プラットフォームdigzyme Spotlight™)のデザイン・・・『こういうふうな機械学習で、こういうモデルをつくったら上手くいくでしょう』という部分を渡来さんたちと一緒に考えて、予算が取れたものを実際に研究員たちに配分して、誰々がどこを作ってくださいね、という感じで開発を進めていきました。
ーー自然と各プロジェクトのリーダーを担うようになっていったそうですね。CTOとしての、採用や教育も。
はい。メンバーが少しずつ増えていく過程で、新入社員へのDRY研究技術の教育も行なっていくようになりましたね。一番最初は、創業直後にアルバイトで入ってもらった礒崎(注6:Principal Investigator、礒崎達大さん)に・・・彼はもともとDRYではなくWET研究の出身だったので、僕と渡来さんでDRYの技術を教えました。
ーーそうだったんですね。DRY技術の教育って、具体的にはどのように進めるものなんですか?
基本的には実例を試すのが一番、だと思っています。プログラミングの勉強って一般的に例えば『1+1=2』みたいな計算をプログラムにやらせたらどうか?というようなことを勉強するんですが・・・
そうするとあんまり身にならないというか、やっぱり『それって現実にどう活かされるんですか?』
いう感じがしちゃうので。
ーーなるほど。
現実の課題を解いていかないとあんまり面白くなくて、身にならないことが多いんですよ。なので、現実的な課題をアサインすることが、教育的にもよいと思っています。
例えば、僕が作っている研修用の資料には、過去に僕自身があたった課題が書いてあります。それを解いて、一通りやってみてもらう、という手法で教育を進めていますね。
さらに最近だと、現実にお客様から頂いた課題をテストケースにして、高山さん(注7:Principal Investigator、高山裕生さん)や礒崎と一緒に解いてもらうやりかたが多いです。
ーー詳しく教えていただきありがとうございます。最近の業務はいかがですか?
個別プロジェクトのマネジメントや、研究リソース管理などが主な仕事になっています。
ーーマネジメントを行うのって、中村さんにとってはどんな感覚ですか?
僕の場合は、マネジメントももともと苦手な項目ではなく、、、学生時代にアルバイトしていたときも、バイトリーダーなどでメンバーの管理などをよくしていたので。そもそも、物事のコアな部分に関わっていないと、気が済まない感覚があるんですよね(笑)
製薬会社勤務のときも、例えば末端で実験・研究だけをしているというよりプロジェクトの詳しい話や本質的なところまで入っていかないと納得がいかず、満足しない方でしたので。
もちろん研究自体は好きなのでやっていたいなという感覚はあるんですが、何も知らずに手を動かすことができないタイプなので・・・
そういう意味でも自然とマネジメントをやっているのかなーという感じがしますね。
でも、これらも礒崎と高山さんに引き継ぎつつあるので、新規技術開発のリードとか、基盤技術の開発がまた僕のコアな業務に戻りつつある、というところですね。
もちろん、DRY・WETのメンバーと、新規技術としてはどういうものを持つべきか?
あるいは、digzymeとしての強みをさらに伸ばすためのアイデアを考えたり深めたりすることは、常々行なっています。
ーーdigzymeとしての強みが、さらに伸びていくためのアイデア、とても気になります。
そうですよね。今後より伸びていくためにという部分に関しては、事業部の皆さんを中心に考えていただいた『事業領域として拡大したい箇所』において必要なソフトウェアだったり、必要なWETの技術などを議論しながら作っていくこともしっかりやっていきたいですね。
それと僕自身は、各プロジェクトのなかで基盤として作ったものがうまく活用されてプロジェクトが進んでいくように動いています。
ーー中村さんは、digzymeで働いていて、どんなところにやりがいを感じますか?
新しい技術を現実の課題に適用しながら、足りない部分を開発したり、アップデートしていったりするところにやりがいを感じます。
例えば、digzyme Spotlight™の開発も、AI、機械学習を用いて酵素を改良しようという試みを実践している存在が世の中でほぼ皆無ななか、行っていました。
それまでも、一般的には酵素の立体構造をみて、『基質と相互作用するタンパク質の場所を変えたら、基質にも影響はあるだろうから変えて、活性をあげましょう』というような研究はされていたんですけれども。
ーー機械学習を用いてという手法は、一般的にとられていなかったわけですね。
はい。それでも、酵素の性能が足りないのでもっとよくしたいというオーダーは多くて、酵素業界全体の課題だったんです。
何かの課題に直面して解決していくという作業が僕は好きなので、
「digzymeだったら、活性を上げる変異体をAIで予測してデザインできそう」
という議論をしながらプログラムを作って、開発していく過程にとてもやりがいを感じました。
ーーSpotlightの開発ケースは相当やりがいに繋がったということですね。
そうですね。
でも、課題そのものは、小さい規模でも大きい規模でも楽しいんですよ。
Spotlightのケースはちょっと大きめですけど・・・日々出てくるちょっとした『これってめんどくさいよね』みたいなものを直して、上手くまわるようになっていく過程自体がとても好きです。
どんな規模でも、新規で技術開発して課題を解決していく『改良』というところにやりがいを感じます。
ーーなるほど。大小問わず課題を解決することにやりがいを感じる中村さんに、digzymeは支えられているんですね!せっかくなので、この流れでSpotlightの独自性についても触れてみたいのですが・・・
開発メンバーが、それぞれの経歴を存分に活かしたからこそ仕上げられたプラットフォームだと耳にしています。この辺りについて、詳しく教えていただけますか。
Spotlightは酵素について、機械学習のアルゴリズムで
「こういう風にやったら、変えるべき場所が予測できるだろう」
というプログラムです。僕は学生時代はもちろん、製薬会社のなかでも機械学習の研究をずっとやっていたので、その知識を活かして。
配列に詳しいメンバーとしては渡来さんや彦有さん(※注8:Informatics Specialist 鈴木彦有さん)。彼らはゲノムとして遺伝子、タンパク質配列を解析するということを研究室でずっとやっていたので。
あとは田村さん(注9:Informatics Specialist 田村 康一さん)ですね。彼は立体構造のデータにすごく詳しいんです。なので、配列と立体構造のデータに詳しい、渡来さん、彦有さん、田村さんの三人に、どういう特徴を学習させたらいいだろうということを考えてもらって、僕の方では『機械学習のモデルにはこういうやり方がありますよね』ということを考えて、最終的には礒崎に実装をしてもらって、仕上がったんです。
ーーまさに『叡知を結集』という感じで感慨深いです。たくさんお聞きできたので、次は仕事で苦労したことや、乗り越えられたキッカケについて伺っても良いですか?
苦労というか、採用活動は結構大変だなと感じながら行なってきました。
会社にとっても、また、採用される個人の人生にとっても、大きいことですしね。重たいことだな、という認識があります。
そんななか、digzymeの未来を担っていただく人材の採用に関して、どう判断したら・・・というところを、かなり悩みながらやってきました。
幾度かの採用活動を経て、最近はようやくコツを掴んだ感じがあります。
渡来さんは、面接時における質問の内容など、採用活動が上手な印象があるので・・・そこは真似させていただいて。
ーーちなみにどんなかたを採用することが多いですか?
やっぱり、話していて違和感がない人・・・これは絶対ですね。
こちらの質問に対して想像の範囲の回答はもちろん、そこを超えた範囲で応えてくださるかたは、前向きに採用したいなという気持ちになります。逆に、思っていたよりも二手三手後ろで止まっている回答をなさる場合は、ちょっと難しいかな、とは思っています。
また、トラブルシューティングが上手な方であることが望ましいです。特にWETの研究は、失敗がつきもの。
DRYはなにかうまくいかなかった時にすぐやり直せますし、僕自身がアイデアを出しやすい分野でもあるのですが、WETの実験はやり直そうと思った時に『また1週間失くなります』・・・など、大幅なスケジュール変更を余儀なくされるわけです。
さらに正直なところ、僕自身がWETにそこまで詳しいわけじゃないので、なにか上手くいかなかった時に『じゃあどうしたらいいと思いますか』と一緒に考えるフェーズにおいて、やっぱり僕以上に詳しくて、ご自身で考えて動けるかたがいらっしゃると嬉しいです。
実験的なトラブルが出ることは本当に多々あるので、しっかり対処できるかという・・・
過去に失敗したケースにどう対応したかなどを伺い、トラブルシューティングが上手な方を採用できるように努めています。
ーーなるほど。教えていただきありがとうございます。
WETのお話が出ましたが、そうしてメンバーを採用してきたからこそ、digzymeのWETの強みって、あれだけすごいんですね。
そうですね。
digzymeのWETに関しては『意外となんでもできる』のが強み、だと思っています。例えば『こういう酵素の評価をしたいです』となったときに、論文を読みながら開発をして、試して、実際に発現させて
評価をして・・・ということをやっていくわけなんですけれど、それって、もちろんちゃんと『研究ができる』方じゃないと難しくて。
例えば、僕が軽く論文読んで追試してくださいって言われて、『実験してみよう!』って軽々できることではないんですよね笑
そこを自然にこなせてるっていうのは、実はものすごくレベルの高いことなんです。
逆にいうと、うちがDRYで解析したものをWETの操作が理由でこれ以上進めません、ということはほぼないです。サラッと言っていますが、これも実はものすごいことです。
ただ、技術はとても高いのですが、リソース的に強いか・・・というところはそうでもなくて、いろんな企業さんとかアカデミアの先生がいらっしゃいますけど、そこと比べるとやっぱり、『特別な微生物株を持っています』とか『特別な遺伝子組み換えが技術があります』とかではなく、あくまで使っているのは公開されているものと同じものを使っているので。
リソース的な強さは正直ないですけど、人員的な研究員としての能力はとても強いと自負しています。
ーーなるほど。心強いですね。
はい。
ちなみにDRYって『因果関係はわからない』ものなんです。どっちが理由で、どっちが結果か?っていうことに関してわからない部分が多いので、僕らが酵素の解析をするときも偽陽性についても考慮しながら進めます。
そこですごく可能性の高いところまで絞っていくのですが、そのあとはWETの技術の高さにとても支えられています。
例えば大腸菌を使うにしても、1株だけじゃなくて何株も用意して、別の生物も色々用意して・・・など、潜り抜けて実験していく技術がとても優れているので、ありがたいです。

ーー今後は、どのようなことにチャレンジしてみたいですか?
基本的には、プロジェクトのステージが進んでいって、上市して何かの役に立ってくれれば、と思っています。
今開発している酵素が、製品になって表に出てくるのは楽しみです。『実はこの製品に入っているんだよ』と言えるくらいになったら嬉しいなと思っていますね。
技術的なチャレンジでいえば、プロジェクトのステージが進んでいったからこその課題が出てくるはずなので、解決していきたいです。
例えば、『こういう化合物を、こういう別の化合物に変化させたい』という時に、『酵素量はこれくらいの量で』『どれくらいの効率で』という数値が現状よりは具体的になってくるはずです。
それを達成できるorできない、が近い未来の重要課題になると予想しています。
さらに次のステージでは“大量生産“が待っています。『製品の価格がいくらなので、そのコストで作るためには、培養液はこれくらいの量で・・・』『この量の酵素が作れないといけない』などの生産性能の目標が見えてくるはずです。
これらの達成は事業継続にも関わる部分ですので、しっかり解決していく予定です。
ーー目標値がもう少し厳しく明確化されていくと、生産性能向上や安定が肝になってくるということですね。他にもなにかチャレンジしたいことはありますか?
完全に人工の酵素の開発はしてみたいですね。
ーー完全に人工の酵素?
『完全に』っていうのは、また難しいですけれども(笑)
普通は天然に存在する微生物が持っている酵素の情報をベースに作るとか、それを改良するというやり方ですが、データからいきなり酵素をデザインするということが、今はAI技術を上手く使うと、少しできるようになってきているんです。
そうすると、もしかしたら何かの微生物の酵素には、ちょっとだけ似ているかもしれないですけど、『ベースの酵素』があるわけではなく、パソコン上でポチポチ作業をしていたら酵素がデザインされて、
それがどの微生物由来でもないもの、というのが作れます。
ーーなるほど。
まぁ、これを例えば食品に入れて、実際に食べたいか?と言われると難しいかもしれないですけども(笑)
今は天然にあるものがベースなのでそこに縛られていますが、それを完全に脱却してオリジナルな酵素を作ることができるので。
実際に何かに使えるかどうかは置いておいても、新しい感じがして面白いですよね。
あとは無細胞系などの微生物に依存しない開発はしてみたいですね。
無細胞系は今社内でもちょっと使っていたり、議論にもなっていたりしますが、基本的にはWETの過程において微生物で遺伝子組み換えして発現・・・という流れが中心なので、やはり『タンパク質が発現しないです』ということが往々にしてあります。というところで無細胞系・・・ーもちろん無細胞でも発現しないことはあるのですがー比較的抑えられることもあるので、そういう意味でトライしてみたいです。 とにかくバイオ特有の不明瞭さがなくなると嬉しいな、と。
ーーバイオ特有の不明瞭さ、というと?
バイオの実験って『なんかよくわからないけど上手くいかない』ということがよくあるんですよね。この『よくわからないけど上手くいかない』が無くなるとすごく嬉しいよね、と思っています。
どうやったら確実にできます、という話では全然ないので、本当に夢の話をしていますって感じなんですけれど(笑)
例えば『培養の条件』一つとっても、この培養条件が一番良いです、ということが理論的にわかるわけじゃなくて、ある微生物を培養する時に、培地にどういう成分をどれくらい入れれば良いか、みたいなことは何度も何度も実験して、この組み合わせが最適っていうのを探し出すんですよね、やり方としては。理論的にこれだ!ってわかるわけじゃなく、なんでかよくわからない・・・。
培養していて、上手く増える時もあれば、そうでないときもある。タンパク質も、作ってみるとあんまり生産量も一定ではなく、多い時もあれば少ない時もある・・・という感じで結構ズレがあるんです、
バイオの実験って。
『失敗する』というのは、極端に『不明瞭なケース』ですけれど、上手くいっているときでも『特別上手くいっているとき』も『微妙だけど上手くいっているとき』もあり、誤差が大きい。
ーーなるほど、それは確かに不明瞭ですね・・・。
そんな不明瞭さのなかでもまず『宿主ごとに上手く発現しない』というのは、やはり大きな課題なので・・・
このあたりが改良できるように、『この世の酵素を、何でも発現できるシステムみたいなものが
あったらいいね』という意味で、あるとしたら無細胞なのかな、という気はしています。
ーー何でも発現。夢がありますね。詳しくありがとうございます。最後に、digzymeに応募を考えている未来の仲間に一言あれば、お願いします。
「バイオの曖昧な課題の解決に最先端技術とアイデアで一緒に挑戦しましょう!」
ーー中村さん、ありがとうございました。
終わりに
▼オリジナル記事はこちら(note)
https://note.com/digzyme/n/n4cb24197110b
「digzyme Custom Enzyme Lab」で期待される実用化例:糖鎖構造構築および難分解性物質へのアプローチ
はじめに
2025年5⽉21⽇(水)、22(木)、23 ⽇(金)
と3日間に渡り開催されたifia JAPAN 2025。
昨年と同様、弊社の代表取締役、渡来が出展者プレゼンテーションを行いました。
その様子をYOUTUBEで公開いたしましたので、ぜひご覧ください。
今回の出展者プレゼンテーションでは、2025年5月21日にローンチされた「digzyme Custom Enzyme Lab」についてご紹介しました。当日は、DRY技術(バイオインフォマティクス解析)とWET技術(実験検証)という二つの技術的アプローチに触れつつ、プラットフォーム全体の概要をお伝えしました。
本記事では、その中で取り上げた『「digzyme Custom Enzyme Lab」で期待される実用化例』2件について、渡来の視点を通じて、各事例における技術的ブレイクスルーやin silico設計の裏側をQ&A方式で詳しくご紹介します。
当日のプレゼンテーションでは全体像のご紹介にとどまりましたが、本記事を通じて、「digzyme Custom Enzyme Lab」の実力と可能性をより具体的にご理解いただける内容となっております。ぜひ最後までご覧ください。
まずは一つ目の事例についてです。

「digzyme Custom Enzyme Lab」で期待される実用化例1
Q.この成果の最も大きな意義は何だと考えていますか?
A.糖質は、構成する糖の結合様式の違いによって物性の差が生まれます。in silico技術で、目的の糖鎖構造を作る酵素を狙って探索ができた例は学術的にも稀で、かつ10個という少数の実験で発見できたことは非常に価値が高いと考えています。
Q.これまでのアプローチと比べて、今回のアプローチは何が革新的だったのでしょうか?
A.本件は、結果的には、当時のAlphaFold2に代表される深層学習(DL)ベースの構造予測技術に対して、弊社独自の詳細な分析技術を適用した点が役に立ちました。これまでのhomologyベースのモデルでは、糖鎖構造を作り分ける微妙なタンパク質構造の違いまでは予測することが困難でしたが、当時のAI技術によりそれらの特徴を一定程度捉えられたと考えています。
(なお、現在の生成モデルを用いた最新のAI技術とはギャップがあるため、本稿では便宜的に「AI」とまとめて表現しております。)
Q.チームや関係者のどのような努力がこの成果につながったと思いますか?
A.担当の研究員が、顧客ニーズを詳しく深堀りして当該酵素のスクリーニング基準をうまく設定し、基盤開発メンバーと連携することで解析プログラムを個別に作成し、この成果につながりました。当社では、既存プラットフォームだけで達成できない課題に対しても、フレキシブルにツール開発を行える点が強みだと思っています。
次に、三菱ケミカル株式会社と共同で行った、二つ目の事例についてです。
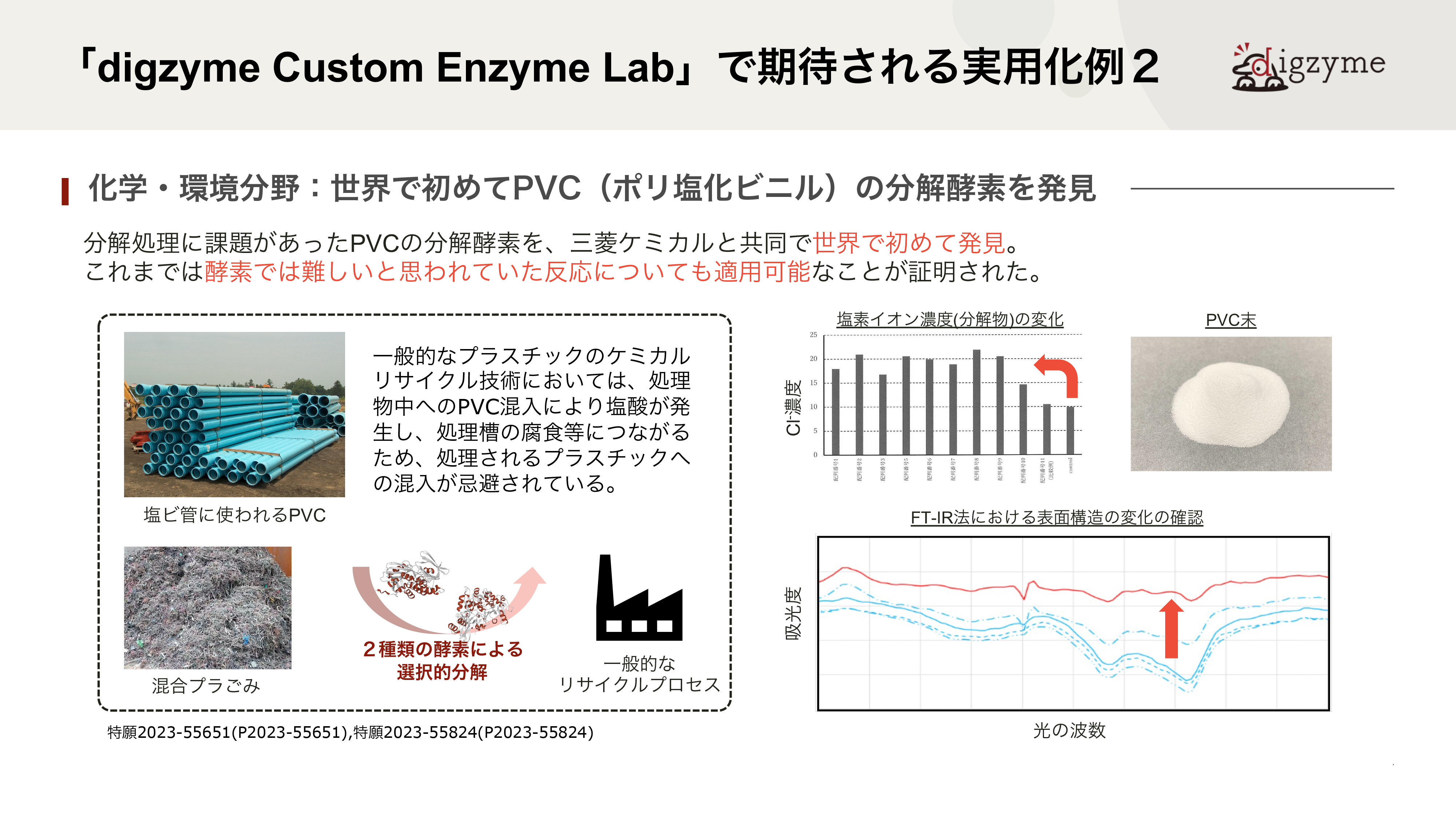
「digzyme Custom Enzyme Lab」で期待される実用化例2
Q.この成果の最も大きな意義は何だと考えていますか?
A.PVCは20世紀から本格的な生産が始まった自然界にない物質で、天然の微生物が進化の過程で分解機構を獲得していないと仮定すると、最適化された酵素は天然からは見つからないはずです。一方で、生物は休眠遺伝子を含めて”最適化されていない”さまざまな遺伝子をゲノムに保有しており、結果として環境変化への適応に活かされるとされています。本件は、その環境適応に寄与しうる酵素をin silicoで人工的に見つける問題とも捉えられるため、難易度が高いテーマでした。
Q.従来、このような酵素を発見するにはどれほどの時間やコストが必要でしたか?
A.近年、人工的なプラスチックの分解酵素を、集積培養に近い形で見出す研究がいくつか見られます。例えば、ある樹脂を海底に一定期間沈めておき、引き上げた後に分解の様子を観察したり、バイオフィルムに含まれる微生物を単離培養したりします。うまくいく場合には、分解酵素を持つ微生物が見つかるので、ゲノム解析やBACライブラリ作成などを通じて分解酵素を同定することが出来ますが、分解が遅いという性質上、どうしても年単位での時間がかかってしまいます。もちろん、分解が観察されず、うまくいかないケースも数多くあると思っています。in silicoでの探索は長くても半年程度で済むため、このような実験時間が長くかかってしまう対象に対してもある程度効果的に用いることが可能です。
終わりに
渡来は、今回の出展者プレゼンテーションを振り返りながら、次のように語っています。
「『digzyme Custom Enzyme Lab』でも、事前の準備期間の中で、これらの共同研究ケースのようなin silicoライブラリを作成して進めていくことができます。高精度のライブラリから精製酵素を試したいお客様におすすめしたいサービスです。」
このコメントが示すように、バイオインフォマティクスを基盤とした酵素設計アプローチは、限られたリソースの中でも実用的な酵素開発を加速しうる可能性を持っています。
今後さらに、酵素の多様な分野への応用が進む中で、「digzyme Custom Enzyme Lab」はその中核を担う技術基盤として重要な役割を果たすと考えられます。