Food Development Expo 2025|CTO Presentation Highlights & Q&A from Our Exhibition Booth
Introduction
Murase from the Food Business Division here.
Digzyme Inc. (hereinafter “our company”) exhibited at Food Development Expo 2025, held from October 15 (Wed) to 17 (Fri), 2025. During the event, our CTO, Mr. Nakamura, gave a talk in the “Exhibitor & Special Presentations” program titled:
“Case Studies of Industrial Enzyme R&D at Digzyme for Developing Differentiated Flavors, Aromas, and Textures.”
We are pleased to share that this presentation has been re-recorded and is now available on YouTube.
In conjunction with our participation in this year’s exhibition and the release of the presentation video, we have compiled a Q&A featuring the topics that were asked most frequently during the event.
This article is designed for those who posed in-depth questions following our “Exhibitor & Special Presentations” session, as well as for visitors who expressed broader interest in digzyme’s industrial enzymes and our development process.
We hope you enjoy reading through the insights we share.
Q1.
By using enzymes designed through digzyme’s in silico technology, what new approaches become possible for addressing challenges in flavor and texture development that were previously difficult to solve?
A1.
Our technology allows us to discover and design new enzymes by enhancing or fine-tuning properties such as activity and substrate selectivity based on existing enzyme characteristics. As a result, it becomes possible to achieve more precise control over flavor-related compound ratios and texture parameters such as viscosity and firmness—areas that have traditionally been difficult to modulate.
Q2.
Besides improving the flavor, aroma, and texture of food ingredients, what other modifications can enzymes enable?
A2.
Enzymes can be used to modify the physical properties of food materials. For example, altering the structure of starch in bread or rice can help prevent hardening over time, and partially breaking down plant proteins can improve solubility.
With conventional enzymes, it is sometimes difficult to achieve sufficient modification under specific materials or conditions. At digzyme, however, we can design enzymes tailored to the desired substrate and environmental conditions, enabling property modifications of materials that were previously challenging.
In addition, we are exploring the use of enzymes to convert or synthesize raw ingredients into food materials with known functional properties.
Q3.
In the future, can in silico enzyme design enhance the sustainability and production efficiency of food development?
A3.
Enzymes optimized through in silico technology can be designed to reduce by-products and to react under lower temperatures or shorter processing times. As a result, it becomes possible to deliver consistent flavors, aromas, and textures while minimizing energy and raw material usage, offering significant benefits in terms of sustainability and efficiency. Furthermore, by applying appropriate enzymatic treatments, by-products or raw materials that were previously discarded can be converted into new products, contributing to the upcycling of food materials.
Q4.
What direction will the development of production strains for efficiently producing enzymes designed with digzyme Moonlight take, and what possibilities could this open up in the future?
A4.
Although we did not cover this in detail during the presentation, we are advancing the development of production strains to enable stable, practical-scale enzyme production. Specifically, this involves selecting microbial strains capable of high-efficiency enzyme expression and optimizing culture conditions for large-scale enzyme production.
In the future, we aim to establish a production strain system that serves as a platform for rapidly commercializing innovative enzymes. This will allow us to provide end-to-end solutions—from designing enzymes tailored to specific needs to bringing them to market efficiently.
Closing Remarks
At Food Development Expo 2025, we received many detailed and insightful questions from visitors, which reaffirmed the growing interest in enzyme design.
We will continue to explore the potential of enzymes together with industry partners. We hope that this Q&A proves helpful and provides inspiration for your future product development and innovation.
Exploring the Treasure Trove of Underutilized Sequence Data: Hikoyu Suzuki on Unlocking Value Through Analysis (Employee Interview)
Introduction
This article is a reprint on our tech blog of an interview originally published on our company’s note in November 2024, made available here so that a wider audience can read it. The content reflects the information as of the time of publication.
(*All organization names and job titles mentioned in the article are those at the time of the interview. In the text, we refer to him as “Hikoyu-san” in line with the internal company usage.)
Informatics × Enzyme Development
Hikoyu Suzuki, who studied molecular evolution at Tokyo Institute of Technology and joined digzyme as a big data analytics expert,
shares his insights in this interview on the frontlines of “data-driven enzyme discovery” from the perspective of research and development.

—To start off, could you tell us about how you came to join digzyme?
Even before I seriously started considering a career change, I had registered with JREC-IN, the career support portal provided by the Japan Science and Technology Agency. By chance, I came across a job posting from digzyme there. Since the company originated from my alma mater, Tokyo Institute of Technology, it felt like a meaningful connection. I also felt that my own skills and know-how could be immediately useful, making me a strong candidate—this combination of factors prompted me to apply.
In my previous role, I worked at a company providing analysis services for biological big data—genomes, transcriptomes, and other “omics” datasets. What was similar to digzyme was that many of the projects were close to bespoke, tailor-made assignments.
It’s similar in format to the collaborative research projects I’ve been involved with at digzyme. Of course, it wasn’t specifically focused on enzyme development at that time.
Analyzing big data requires computational expertise, so it’s not something just anyone can do. My work involved handling projects from companies in the food and pharmaceutical sectors, performing analyses tailored to each case, and providing some form of conclusion or insight. You could think of it as being somewhat like consulting work.
—So, in your work, were you combining R&D consulting with biological big data?
Yes, that’s right. I spent about five years at such a venture. A clear example of the projects I handled would be related to antibody therapeutics. Since antibody drugs now occupy the top ranks of pharmaceutical company sales, we often worked on analyses connected to them.
An “antibody” is a protein that can bind specifically to a particular antigen (target). Antibody therapeutics therefore tend to have fewer side effects than conventional drugs and can be highly effective in treating cancers or rare diseases. But antibodies are, of course, proteins, meaning there is a base “human antibody gene.”
The process involves inserting these antibody genes into cultured hamster cells, letting them produce the antibodies in large quantities, purifying them, and turning them into a product. Along the way, you need to consider things like how to optimize the culture methods to produce efficiently at scale, or what sequence to design if you’re developing a new antibody drug.
Nowadays, gaining insights from big data is often part of these considerations. For example, something that works at a small experimental scale doesn’t scale linearly to full production. Scaling up 100 times doesn’t mean you can just produce 100 times as much (laughs). In such cases, we analyze what’s happening by obtaining and studying biological big data, and that’s the kind of support I provided.
—I see. Nakamura-san also mentioned that controlling biological systems at production scale is challenging. What kind of work have you been involved in since joining digzyme?
Right after joining, my work was mainly focused on development based on the concept of synthetic biology, specifically on programs for exploring reaction pathways—including novel reactions—to enable organisms to synthesize target compounds.
For example, if you want to produce a target compound D from a starting compound A, you might need to go through B and C along the way. Because multiple steps are often required, you first need to predict the intermediate reaction pathways; without this, you can’t search for enzymes to catalyze those reactions.
In synthetic biology, when using cultured cells or microbes to produce something useful, a single reaction is rarely sufficient, so predicting the reaction pathway is essential. My role was to explore optimal pathways among many possible options.
Nowadays at digzyme, we mainly handle industrial enzymes, so we don’t need to worry about reaction pathways anymore. We just need to provide the enzymes that meet the customer’s requirements. As a result, my focus has shifted to developing and maintaining libraries of enzyme variants so we can supply
ChatGPT:
—Thank you for touching on the library development! Could you also tell us about your recent work?
I develop and maintain analysis environments that can be used even by WET lab members who aren’t familiar with DRY (computational) analysis. Specifically, this is what’s generally referred to as introducing an IDE (Integrated Development Environment).
For beginners, just installing analysis software can be a major hurdle. Many people might imagine “just download the installer, double-click, and it’s on your computer,” but in bioinformatics, most commonly used tools don’t work that way. From the installation stage, everything is done via the command line—typing commands to instruct the computer—so beginners often get stuck right at this step if they don’t already have some technical knowledge.
To address this, we provide pre-configured environments that remove these obstacles. Introducing an IDE is essentially this solution.
Additionally, many bioinformatics tools don’t have a GUI (Graphical User Interface), which makes them even less approachable. To make them as intuitive as possible, we leverage Jupyter Notebook.
Here’s how it works: WET researchers typically create “lab notebooks” to record experiments. In the same way, we create “analysis notebooks.” These aren’t just simple notes—they contain the executable commands, and even buttons to run those commands. This setup is useful when you want to repeat similar command-line procedures for another project.
In extreme terms, you can perform analyses without fully understanding the underlying programs being used—though of course, it’s not advisable to use it without any understanding at all (laughs).
I work on these environments in collaboration with specialists like Tamura-san (Informatics Specialist, Koichi Tamura) and Isozaki-san (Principal Investigator, Tatsuhiro Isozaki).
—I see. By the way, in what kinds of situations do WET lab members actually perform DRY (computational) analyses?
The main scenario is typically the phase after signing an NDA with a client, when we need to assess feasibility.
Ideally, WET researchers can perform quick analyses during meetings with the client to evaluate feasibility on the spot. This way, even if a DRY team member isn’t present, they can provide immediate, informed feedback, which makes the process very smooth.
Additionally, in the phase of advancing R&D as an actual business project, being able to make decisions about experimental targets and strategies based solely on DRY analysis results—without constant support from computational specialists—offers a significant advantage in terms of development speed.
—Thank you for explaining. What other kinds of work are you involved in?
I’m also involved in handling actual analysis and investigation tasks for individual projects, as well as developing new analysis algorithms. This includes reading the latest research papers and implementing improvements to enhance the accuracy of our existing internal tools. On the biological side, I sometimes work on calculating metrics used for decision-making or assessments.
—What kinds of papers do you usually read?
My background is in molecular biology and molecular evolution, so I’m particularly skilled at analyses involving protein amino acid sequences, as well as “quantitative” information linked to sequences, such as genomes and transcriptomes. As a result, I often read papers that deal directly with gene sequences.
Within the “notebooks” I mentioned earlier, I examine these papers to see if any of the sequence data processing methods can be effectively applied to our analyses.
—While Tamura-san, another informatics specialist, focuses on the structural aspects, Hikoyu-san, you advance analyses daily from the perspective of gene sequences, correct?
That’s right. Our areas of expertise are nicely complementary, so at digzyme we can analyze enzymes from multiple perspectives. I think that’s one of our key strengths.
—Next, could you tell us what aspects of working at digzyme you find most rewarding?
What I find most rewarding is that the results we develop are highly likely to be used directly as a means to create new value for our clients and generate profit for the company.
Many of our clients continue to work with us long after the initial PoC, which serves as a “test of whether we can meet real-world needs.” I can’t share many details due to confidentiality, but I feel that clients recognize the significant synergy they gain from working with digzyme.
Going forward, I want to continue providing new value in response to client needs and make meaningful contributions.
—Hikoyu-san, how do you view the provision of new value that is uniquely possible at digzyme?
As I mentioned earlier, as a premise, there is already a vast amount of genetic information related to enzymes available as big data. Among this data, many sequences have been determined, but their functions are not well understood.
Just because a sequence exists doesn’t mean it lacks function—it’s naturally occurring, and there’s a high likelihood that many of these enzymes can catalyze reactions useful to humanity. How do we find them? Conducting experiments one by one would be impossible. Instead, we first narrow down the search computationally—identifying “this area looks promising”—and of course, experiments are eventually necessary, but this approach maximizes the probability of success with minimal experimental effort. That initial filtering is extremely important, and I believe it’s the essence of digzyme’s technology.
In line with our mission at the time—“Let’s go meet the enzymes that will change the world” (as of November 2024; now updated to “Catalyzing breakthrough options in manufacturing.
Creating lasting vitality for people and Earth.”)—we want to continue uncovering the treasure trove hidden within big data and bring those enzymes to light.
—Thank you for also touching on the essence of digzyme’s technology and your stated mission!
By the way, could you explain the background as to why so many sequences have been recorded in big data but their functions remain poorly understood?
Originally, the practice began with individual researchers registering the sequences of the genes they were studying, so that later, others could verify them or reuse the data in different studies. However, the situation changed drastically as DNA sequencing technology improved.
Especially from the 2010s onward, the widespread adoption of next-generation sequencers made it possible to cheaply decode massive amounts of DNA sequences. Previously, researchers would typically target specific genes using PCR and then sequence them, but now it became feasible to “just read the whole genome!” or “sequence all the RNA being transcribed!”—essentially, a “sequence everything” approach.
As a result, huge volumes of sequence data started accumulating continuously across a wide variety of organisms. While this data has high potential for reuse in other research, the researchers who generated it usually only examine the parts relevant to their own studies.
On the other hand, researchers who have been active for a long time often notice that data collected in other studies can be valuable from the perspective of their own work. This insight led digzyme to adopt the idea of focusing on the reusability of data from a researcher’s point of view. Going forward, we aim to continue uncovering data that holds the potential to address unexplored future needs, mining valuable information that is still hidden.
—I see, that really helps me understand the background. Hikoyu-san, you have such a wide range of expertise, but could you tell us more about what you specialized in at university?
During my student years, I conducted research on evolution under Prof. Norihiro Okada (Emeritus Professor, Tokyo Institute of Technology) and Prof. Masato Nikaido (Associate Professor, Tokyo University of Science, formerly Tokyo Institute of Technology). Specifically, I analyzed molecular-level data such as genes and proteins to investigate evolutionary relationships among different organisms and, from there, explored how genes evolve and the functions of ancestral genes.
At that time, Okada Lab focused on cichlid fishes in the three major lakes of East Africa—Lake Victoria, Lake Malawi, and Lake Tanganyika. These freshwater fishes are highly diverse morphologically and ecologically, and each lake hosts many endemic species, making them a globally recognized model for studying evolution and speciation. The lab energetically pursued research aiming to explain the enormous diversity of cichlids through their genes.
Around the same time, the team led by Prof. Yohei Terai (Associate Professor, SOKENDAI) proposed the sensory drive hypothesis, suggesting that evolution of sensory genes—such as those involved in vision—can promote speciation. Inspired by this, Prof. Nikaido’s team examined other sensory systems, focusing on olfaction.
I specifically focused on olfactory receptor genes, which detect diverse odor molecules and transmit signals to cells. In fish, the olfactory rosette—a petal-like structure visible to the naked eye when the nostrils are opened—houses numerous olfactory neurons that enable fish to sense odors in water. Each of these neurons is believed to express a different olfactory receptor gene. My WET experiments involved classifying these genes phylogenetically, visualizing the neurons expressing each gene, and using olfactory marker proteins to map the neurons expressing these genes.
Over time, my research shifted slightly from cichlids to the evolution of olfactory receptor genes themselves. For instance, Prof. Nikaido had discovered that while most vertebrates possess a single olfactory marker protein gene, teleost fishes have two. I investigated the expression of these two genes in detail and found that they are expressed in different olfactory neurons, and one is also expressed in horizontal cells of the retina—revealing that a gene previously thought to be exclusive to olfactory neurons is also used in retinal neurons.
Additionally, when analyzing the coelacanth genome, I discovered that several olfactory receptor gene lineages previously thought unique to terrestrial vertebrates are also present in “ancient fishes” like coelacanths and polypterids. I also identified a previously unknown type I pheromone receptor gene (ancV1R) conserved across vertebrates, including ancient fishes. The discovery of ancV1R was published in a paper and announced in a press release by Tokyo Tech, and it remains one of my most cherished research achievements.

At some point, I also began performing analyses using data generated by next-generation sequencers—devices capable of decoding hundreds of billions to a trillion DNA bases in a short time. There were a few triggers for this shift.
Originally, Okada Lab frequently used analyses of genomes or individual gene sequences. Several assistant professors and postdocs in the lab had self-taught bioinformatics skills and were already applying them to their research, so I thought, “Why not give it a try myself?”
I had some early experience with computer programming as a child, having played around with BASIC, so I started using Perl, a language with similar syntax. Although Perl is less commonly used today, at the time it was widely employed in web application development and excelled at string processing, making it ideal for handling DNA and amino acid sequence data in bioinformatics.
Through this work, I became recognized in the lab as a “student who can do bioinformatics.” Later, Okada Lab’s proposal for a Ministry of Education, Culture, Sports, Science and Technology (MEXT) New Academic Field research project titled “Genetic Basis of Complex Adaptive Trait Evolution” (2010–2015), led by Prof. Mitsuyasu Hasebe (National Institute for Basic Biology), was accepted. Thanks to the recommendations of Prof. Okada and Prof. Nikaido, I was fortunate to participate in the project as a student skilled in bioinformatics.
This project was a challenging endeavor aimed at unraveling aspects of complex evolutionary phenomena across organisms by actively acquiring and utilizing biological big data such as genomes and transcriptomes, leveraging next-generation sequencers, which were just becoming widely adopted at the time.
At the same time, I believe there was an educational aspect, as young researchers like me learned bioinformatics skills, including how to handle next-generation sequencing data. Participating in this project allowed me to gain invaluable experience and skills, which I have been able to apply in both my previous work and my current role at digzyme.
—I see, thank you for explaining that in detail. Hikoyu-san, having been deeply involved in both WET and DRY work, what kind of challenges or new initiatives would you like to pursue at digzyme moving forward?
Exactly—it's about cultivating “dual-skilled” talent in both WET and DRY. When WET researchers gain familiarity with DRY approaches, it can accelerate research, and when DRY researchers understand WET, they can develop solutions that better fit the life sciences environment.
In fact, in my previous position, I was invited as an instructor to give hands-on bioinformatics seminars, explaining techniques to both industry and academic audiences. So at digzyme, I envision something similar—internal seminars or training sessions that help bridge WET and DRY knowledge within the company.
Creating the IDE was also a stepping stone. Of course, simply using the IDE doesn’t automatically build expertise, but moving forward, when new WET members join, we could hold a seminar as a first step—something like, “Let’s try an analysis using the IDE!” I hope to coordinate with CTO Nakamura-san to make this happen.
—Exactly. Because many of digzyme’s DRY engineers come from biological backgrounds, this kind of education is uniquely possible here. In that sense, the composition of our team is indeed a rare and valuable asset.
Exactly. At digzyme, we actually don’t hire people purely from information science backgrounds, because developing enzymes requires deep biological knowledge.
Everyone on our core team—Watari-san, Nakamura-san, Tamura-san, and myself—comes from a biology-related field and then built up our knowledge of information science from that perspective.
Since WET researchers already have a strong background in biology, they have the potential to take the same path—learning DRY techniques from a biological starting point—and digzyme is uniquely positioned to support that kind of training.
Ideally, this type of talent development should be a national policy priority in Japan and addressed across the industry. But in practice, it’s quite challenging for WET researchers to learn DRY skills. The main reason is that opportunities to do so are rare. Unless you’re in an environment like digzyme—where progress depends on understanding both biology and information science—it’s difficult to gain the necessary experience.
—Is there an aspect that makes it highly dependent on the environment?
—There’s also that aspect. In fact, I’ve even heard that in experiment-focused labs, if someone just sits at a computer typing away without doing any wet experiments, they might be seen as “slacking off.”
But in reality, while it’s understandable that people from pure information science backgrounds might avoid studying biology, for biologists, you really can’t separate the two. Now that it’s standard to sequence massive amounts of DNA with next-generation sequencers, avoiding computational approaches puts biologists at a clear disadvantage.
Some might think that the DRY part can just be left to collaborators, but that ends up making the bioinformatics experts extremely busy (laughs). Ideally, even in academia, researchers who study a particular organism should also be able to perform DRY analyses themselves. I believe the core members of the New Academic Fields project I mentioned earlier shared that same ideal, and I think our CEO, Watari-san, would agree as well.
—Finally, do you have a message for future colleagues who are considering applying to digzyme?
—At digzyme, you’ll find an environment where you can work while nurturing both your own curiosity and your desire to contribute to others.
—Thank you very much, Hikoyu-san.
Closing Remarks
▼ Original article is available here (note)
https://note.com/digzyme/n/n7fbee270334b
Coordinating Research and Social Implementation: Isozaki on Tackling Challenges in the Field (Employee Interview)
Introduction
This article was originally published on our company’s note(Japanese only) in October 2024. To reach a wider audience, we are also reposting it here on our official tech blog. The content reflects the information available at the time of publication.
(All organizational names and job titles mentioned in the article are as of the time of the interview.)
ーーFollowing our first interview with our CTO, Mr. Nakamura, this time we are interviewing Mr. Isozaki, our Principal Investigator (PI). Thank you very much for joining us today, Mr. Isozaki.
To begin, could you tell us how you came to join digzyme? During your student days, you moved from the Tokyo Institute of Technology to the doctoral program at Kyoto University, correct? And after that, you joined digzyme.
Yes. I actually left the doctoral program at Kyoto University before completion. When I returned to Tokyo afterward, I found out that our CEO, Naoki Watarai — who had been my senior by one year at Tokyo Tech — had just founded digzyme. I thought it sounded really interesting, so I reached out to him, and that was the beginning.
ーーBack in your Tokyo Tech days, what kind of relationship did you have with our CEO, Naoki Watarai?
I originally met Naoki Watarai back at Tokyo Tech, when we were both involved in iGEM.(※The International Genetically Engineered Machine Competition)
ーーiGEM!digzyme is also supporting it as a sponsor.
Yes. iGEM is the world’s largest synthetic biology competition, where even undergraduates can engage in realistic research activities without joining a lab. During iGEM, Watarai-san and I were on the same team representing Tokyo Tech. It was a small team of just three people, so we got along quite well from the start. I could already tell back then that Watarai-san was an incredibly powerful and remarkable person. Over time, I became convinced that 'if Watarai-san starts a company, it’s going to be something really interesting.'
At the time, my own field of study didn’t give me deep expertise in enzymes, so I wasn’t that knowledgeable about them yet. After returning to Tokyo, I spent a while working in technical sales at another company. But after a short period of side work, I eventually joined digzyme full-time.
In my technical sales role, I would explain the platform we were handling to customers, then bring the information back to the company and hand it over to the appropriate engineers. That itself is an important job, and in fact it’s a position that digzyme still needs. However, most of the work was focused on customer negotiations, and I had very little direct involvement with new technical developments. I sometimes felt a sense of powerlessness in that kind of bridging role.
That experience was very valuable in giving me a hands-on understanding of what technical sales is like. But as a researcher, I believe creativity comes from being able to set your own challenges and run toward your own goals. At digzyme, I felt I could do just that, which is why I decided to commit fully. And I’m really happy that I can actually work this way now. I get a great sense of accomplishment tackling daily challenges.
ーーI see. It’s great to hear that you’re moving in the direction you want, and I feel happy listening to that! Specifically, what kinds of tasks or aspects do you find particularly rewarding?
I develop analytical technologies every day to solve challenges in collaborative research and new business projects. What I find particularly rewarding is the part where we discuss how to effectively translate biological features into dry analysis and then actually test them in practice. Specifically, we take the 'abstract challenges' we receive from our clients and break them down into very detailed hypotheses about the biological characteristics — testing them in dry analysis, and eventually connecting them to wet experiments. We refer to this process as 'translating' the features.
The procedure usually starts by identifying which aspects of biological features are likely to contribute to the problem we want to solve. In dry analysis, we formulate hypotheses and analyze things like structural differences, patterns in temporary sequence motifs, or whether the protein can be expressed in a particular species. But it doesn’t stop there: we also validate these hypotheses in the lab through wet experiments, or, in the case of collaborative research, have our partners test them directly.
Whether it’s collaborative research or our own projects, we can perform the full cycle of hypothesis testing — in other words, we can actually prove the hypotheses we’ve worked hard to develop. That is what gives me the greatest sense of fulfillment.
At digzyme, DRY and WET are seamlessly connected. Because these two foundations are solidly in place, it makes for a really excellent environment.
ーーNow, Isozaki-san, you are well-versed in both DRY and WET approaches… In your daily work, are there moments when you feel that your past experiences have been particularly helpful?
Actually, when it comes to DRY, my experience as a student was really just as a user — I could use some commercial analysis tools a little bit. I wasn’t able to write code or anything like that…
ーーWow! I can’t believe it…!
That’s right. And on top of that, I didn’t really know the logic behind how DRY tools are made — I was really just ‘able to use them.’ So I don’t think you could have called me a ‘DRY person’ back then (laughs). I was mainly a WET person.
During my doctoral program, I went offshore and studied marine bacteria. My main work was WET, and I only used Linux a little as a supplementary tool.
I used techniques like RNA-Seq — one of the next-generation sequencing (NGS) methods, which allows comprehensive detection of gene expression, known as the 'transcriptome' — to estimate the infection targets of marine viruses based on their genomic genes.
ーーI see. How about during your master’s program?
Up until my master’s program, I didn’t even know Linux, and I only used Windows — basically just Excel (laughs). At that time, I was even further away from genomics. During my master’s, I worked on cancer models using mice: transplanting cancer cells into mice, collecting samples, preparing tumor sections — it was almost like craftsmanship — and observing them under the microscope. You could say it was somewhat similar to what a pathologist does with human samples.
However, as I started attending academic conferences, I realized that if I couldn’t do any DRY work at all, I wouldn’t be able to survive, whether in academia or in any other research role.
I think many people who specialize in WET share this feeling. Most of the current members at digzyme would probably say the same thing: 'If possible, I really want to learn this too.
ーーBeing able to understand both DRY and WET approaches seems to have a lot of advantages.
Yes. But often, it can feel like you’re just pending, not really sure what to do. That’s part of the reason I deliberately changed labs when moving from my master’s to my doctoral program. I moved to a lab where I could potentially work with both WET and DRY. That said, as I mentioned earlier, I was only using commercial tools at the time, so I hadn’t even touched Python in terms of coding until I joined digzyme.
At digzyme, I learned everything from Watarai-san and Nakamura-san. Back then, we would occasionally hold programming sessions at a certain family restaurant (laughs), where they’d give me homework and I’d gradually improve.
Interestingly, having WET experience actually benefits DRY development. Understanding how WET researchers feel when using DRY tools is really valuable. Since we’re here, I can also explain what it means to do something in DRY that’s normally done in WET. First of all, you obviously can’t perform the experimental part of WET in DRY. What you can do in DRY is the investigative part. For example, at digzyme, 'enzyme discovery' is exactly that. Deciding which sequences to propose for a given challenge is normally part of the broader 'researcher' role. But because digzyme has the two foundations of DRY and WET, this 'enzyme discovery' is handled by DRY specialists.
If you can automate and quickly judge what sequences are likely to be effective in DRY, experiments in WET can proceed immediately afterward, and the number of experiments can also be reduced — there are many benefits. The same applies to 'enzyme modification.' You can quickly see in DRY where modifications should be made. But originally, all of this is part of the broader 'researcher' role. So, having WET experience naturally contributes to DRY development.
ーーGot it! That makes sense, thanks.
Going back a bit, could you tell us more about your research on marine viruses during your PhD?
I’ve always wanted to study viruses that exist in unusual places, not just in the ocean. As I mentioned earlier, I chose a lab where I could also do some DRY work, which led me to the Marine Molecular Microbiology Lab.
The motivation for wanting to study viruses in unusual environments goes back to when I was in elementary school. At that time, there was a somewhat popular book called The FUTURE is WILD (by Dougal Dixon and John Adams, with contributions by Takayuki Matsui and Akiko Tsuchiya), which simulated what kinds of creatures might thrive on Earth after humanity is gone. The book included CGI depictions of fantastical future creatures, like giant squid that looked almost like elephants — it was really captivating.
I was also interested in paleontology books. In evolution, when you trace ancient organisms, they generally get smaller — starting from microbes and gradually getting bigger is quite different from the world we live in now. I found that monster-like, imaginative aspect really fun (laughs).
So, I could have gone into a field like 'extremophile microbiology,' but I was drawn to something a bit more unusual. For example, there’s a hypothesis that viruses might have contributed to the development of mammals. The genes responsible for forming the placenta are composed of retroviruses, and in fact, the reason this suddenly appeared can be explained by viral evolution. Studying particularly unusual viruses like these seemed fascinating, and that’s what influenced my choice of research direction.
ーーI’m also curious about what viruses really are — they’re quite fascinating. Now, changing the topic a bit, could you tell us about any challenges you’ve faced in your work so far?
The main challenge is translating the abstract requests from clients into 'analytical approaches that are both commercially valuable and feasible.' Often, clients don’t know how detailed their requests should be for digzyme, or they simply can’t specify the level of detail, which makes the requests quite abstract.
For example, they might say something like, 'We don’t even have a concrete chemical structure yet, but we want something with roughly these properties,' or, 'It would be great if the production of this current product could be increased — we’re not sure whether an enzyme can be used or not.' When addressing these kinds of requests, we have to compare conventional methods and competitors with digzyme’s unique technologies, assess whether we truly have an advantage, and also consider market fit and other perspectives. Turning all of this into a concrete analytical approach is a challenge each time.
As for how we overcome this, we resolve it by consulting the right people, both inside and outside the company, as early as possible. Also, now that the business team has grown, I’m less often involved in the initial client meetings, so I personally encounter these abstract requests less frequently. I’m grateful that, as a company, the layer that converts abstract challenges into concrete plans has been steadily strengthened.
ーーWith the team growing, it seems that the environment for solving challenges is steadily improving. By the way, speaking of 'consulting the right people inside and outside the company,' Isozaki-san, I get the impression that you communicate very actively within the company. I personally benefit from that all the time, but do you do the same externally as well?
Yes. For new business projects, I often interview experts using platforms like VisasQ. In particular, for topics related to the Food Business Division, we sometimes gather experts through Mr. Takuo Miyauchi (Director and CSMO) and get their insights.
ーーThat’s reassuring. Are there any insights from these interviews that have particularly stuck with you?
There are many, but as one example… When we asked whether it would be commercially viable if the activity of a certain enzyme (the kind used in tests) could be greatly increased, we received the following comment:
‘The activity of this enzyme has never increased beyond a certain point. Of course, if it could be increased, the accuracy of the tests would improve, which would be good… but I’ve never seen such a case from a technical standpoint.’
ーー‘I’ve never seen anything like that!’ — That must have been really thrilling!
Yes. I could confidently tell myself, ‘This will really hit the market.
ーーThank you for sharing such a wonderful example. I have a follow-up question: what principles or values do you prioritize in your work?
For me, it’s about keeping open communication both inside and outside the company. To achieve this, I focus on building relationships where it’s easy to consult each other early when problems arise.
When psychological barriers are low, it’s less likely for relationships to turn sour. I’m careful not to signal any ‘walls’ to others, because visible barriers can create an ‘us versus them’ mindset. As long as the relationship is at least neutral, it prevents unnecessary conflict and makes problems less likely to occur.
ーーThat’s quite moving. Speaking of Isozaki-san, what kind of challenges would you like to take on in the future?

What I am currently working on is the overall management of WET research operations.
In the past, employees were organized flatly under our CTO, Nakamura. However, starting this year, Takayama-san (PI = Principal Investigator, Hiroo Takayama) and I have taken on a layer above all WET members as PIs. Under us are the PMs (Project Managers), while DRY members report directly to Nakamura.
Since I’ve never directly managed the research work of WET members before, it has been a bit challenging. I am moving forward with support from Takayama-san, my fellow PI.
Looking ahead, I hope digzyme will strengthen its PM layer even further. Ideally, we want to increase the number of WET members who have some understanding of upstream (DRY) work.
That said, I realize that diving straight into DRY work can be confusing—people might not know exactly what to learn at first. That’s why I plan to convey this gradually through on-the-job training (OJT).
ーーI see. If the number of WET members capable of DRY analysis continues to grow, the organization will become even stronger. Are there any other challenges you would like to take on?
I want to promote the development of analysis pipelines that reflect the characteristics of enzyme families based on their three-dimensional structures. By doing so, we can tap into unmet needs across the enzyme industry and explore novel approaches that haven’t been tried before, which should lead to greater diversification and improved productivity in enzyme-based manufacturing. I hope to advance this together with Mr. Koichi Tamura, our informatics specialist.
digzyme’s strength lies in our ability to handle both upstream and downstream processes, while also preparing for production. This is fundamentally different from the nuance of “AI doing a bit of analysis for you.
ーーIt seems like this will become a move that truly leverages our strengths.
Yes. I think digzyme’s biggest distinguishing feature is that it’s deep tech with a genuine market-in mindset. In particular, all the executives naturally think in terms of scaling.
—Of course, the fact that we can differentiate ourselves in specific technologies comes from having professionals from various fields gathered here—but even putting aside whether it’s technically feasible right now, we have our antennas up for ideas that could significantly propel the business forward. That’s why we have this virtuous cycle: we can define the necessary technologies and reliably develop them.
In some cases, deep tech companies aren’t focused on scaling. But our philosophy is that a vague notion like “it’s fine as long as it makes some money” or “this technology is amazing, so surely everyone will want it” just won’t cut it. No matter how impressive a technology is, if it’s for applications people don’t actually want, it can easily end with a “Sure, it’s innovative, but…” and go nowhere.
Because we have a market-in mindset, we can truly move our customers. That’s why it’s so important to have people like Mr. Watarai leading the charge—investigating the cutting edge of business with scaling in mind, and translating those insights into precise technical language.
ーーFinally, could you share a few words for those future colleagues who are considering applying to digzyme?
We have a diverse team in terms of both personality and research or professional backgrounds, offering opportunities to gain experience across a wide range of fields beyond just research. So, if you’re eager to take on new challenges, you’re very welcome here!
▼Original Post[note(Japanese only)]
https://note.com/digzyme/n/n4cb24197110b
Building Core Technologies and Talent to Bridge Discovery, Design, and Expression: From the Perspective of CTO Nakamura (Employee Interview)
Introduction
This article is a reprint of an interview originally published on our company’s note page in September 2024. To make it accessible to a wider audience, we are sharing it here on our official tech blog. Please note that the content reflects the context and information available at the time of the original publication.
— This marks the very first interview in our series! Thank you for joining us, Nakamura-san.
To begin, could you tell us how you joined the company? …Although, come to think of it, you’re actually one of the founding members, aren’t you?
Yes, that’s right.
I co-founded digzyme Inc. while I was still a student at Tokyo Institute of Technology, together with Mr. Torai¹, who is now our CEO, and Professor Yamada², who serves as our CSO and is an associate professor at Tokyo Tech.
The inspiration for starting the company came from research we were conducting in the Yamada Lab³, which later became the foundation of digzyme. We saw strong potential for turning that research into a viable business.
— The research that became the foundation of the company… we’d love to hear more about it. What aspects made you see its potential as a business?
It was research that eventually evolved into what is now digzyme Moonlight™, focused on enzyme discovery.
At the time, Professor Yamada and Nagase & Co., Ltd. were conducting a joint research project. I joined them and contributed by performing additional computational analyses.
Specifically, Nagase approached us with a request:
"We’re looking for an enzyme with these specific properties."
Our task was to search for suitable enzymes from a database based on that request. The process was going well—we were consistently identifying promising candidates. So, the initial plan was to publish the results as a research paper.
From there, I started thinking: What if this discovery technology could be applied to other domains as well?
That line of thinking gradually led us to explore broader possibilities—and that was the starting point for digzyme’s business model.
In terms of its broader applications, what I specifically had in mind was this:
Could we identify enzymes capable of catalyzing synthetic reactions for compounds where no such enzymes have been discovered yet?
That’s exactly what we achieved through our collaboration with Nagase.
The project focused on enzymes from a particular plant that produces a unique compound. But extracting the enzyme by physically grinding the plant each time? That’s obviously not scalable.
So we thought: What if we could reproduce this biosynthesis using microbial enzymes instead?
We began screening for enzymes that might be able to catalyze the same reaction—and we found quite a few.
In fact, we identified a number of enzymes that could potentially synthesize previously unknown or commercially unavailable compounds. That was really exciting.
— That’s incredible!
Yes, and from there, we began to discuss in the paper the possibility that these enzymes might allow us to synthesize compounds that were previously considered extremely difficult to produce—with much more ease than expected.
When I reported this to Professor Yamada, he pointed out that there must be many people out there who need exactly these kinds of enzymes. That led to the idea that this could be really interesting as a business.
And the timing couldn’t have been better—around that same time, Mr. Torai was actively exploring the idea of launching a startup.
So with that momentum, the three of us—Professor Yamada, Mr. Torai, and myself—decided to start working together.
Once we did, we immediately began receiving inquiries from people saying,
"If there’s a technology that can identify enzymes like that, we’d love to use it."
That strong interest convinced us: there’s real demand out there—so we decided to found the company.
ーー Indeed, conventional enzyme development has required an enormous amount of trial and error to identify genes encoding enzymes suited to specific purposes.
Because of this, there has been a heavy reliance on serendipitous discoveries, which brings uncertainty and results in enormous development costs. Considering these challenges, it’s clear there is substantial demand for better methods.
After founding the company, what kinds of work have you been involved in?
Right after the company was founded, I worked on developing the core technology — the enzyme discovery software. At the same time, we began exploring how to further apply this technology to develop areas that would become key strengths for the company.
We already had several research and development themes internally, so I was also involved in those development projects.
However, I was still a student for the first six months after founding the company. After graduation, I worked at a pharmaceutical company for nearly two years.
During that period, I participated in discussions around technology development while balancing this work alongside my role at digzyme.
— After that, you transitioned to committing full-time to digzyme, correct?
Yes. Around the time I fully committed to digzyme, a project funded by NEDO⁴ (New Energy and Industrial Technology Development Organization) started.
In this project, we worked on designing spotlight⁵ (digzyme Spotlight™, our enzyme function improvement platform). Together with Mr. Torai and the team, we discussed ideas such as:
“If we build a machine learning model like this, it should work well.”
Once the budget was secured, we allocated resources to the researchers accordingly, directing who should develop which parts, and proceeded with the development in that manner.
— Naturally, you gradually took on the role of leader for each project. You also became responsible for recruitment and training as CTO, correct?
Yes. As the team gradually grew, I also began training new employees in dry lab research techniques.
The very first was Mr. Isozaki⁶, who joined us as a part-time staff member just after the company was founded. Since he originally came from a wet lab background rather than dry lab, Mr. Torai and I taught him the dry lab techniques.
— I see. So how do you actually go about training people in dry lab techniques?
Basically, I believe the best way is to work through real examples.
When people study programming in general, they often start with simple exercises like “what happens if you program 1 + 1 = 2?” But…
that kind of practice doesn’t really stick, and it often leaves people wondering, “How does this actually apply in real life?”
— That makes sense.
If you don’t tackle real-world problems, it’s often not very engaging and doesn’t translate well into practical skills. So I think assigning realistic tasks is crucial for effective learning.
For example, the training materials I create include problems I personally worked on in the past. Trainees solve these problems and go through the entire process as a way to learn.
More recently, we often use actual challenges from our clients as test cases, and have trainees work through them together with Mr. Takayama⁷ and Mr. Isozaki.
— Thank you for the detailed explanation. How have your recent responsibilities been?ですか?
My main responsibilities have been managing individual projects and overseeing research resource allocation.
— How do you feel about taking on management responsibilities?
For me, management has never really been something I struggled with. Even when I was a student working part-time, I often took on roles like team leader, managing members and coordinating tasks. I have this feeling that I can’t be satisfied unless I’m involved in the core aspects of things (laughs).
When I worked at the pharmaceutical company, I wasn’t the type to be content just doing experiments or research at the ground level—I needed to dive into the detailed discussions and the essence of the projects to feel satisfied.
Of course, I enjoy the research itself and want to keep doing it, but I’m not someone who can just work blindly without fully understanding what’s going on…
In that sense, I guess naturally I ended up taking on management roles.
That said, I’ve been gradually handing over these management responsibilities to Mr. Isozaki and Mr. Takayama, and I’m now returning more to my core work—leading new technology development and building foundational technologies.
Of course, I’m always thinking about what kind of new technologies our dry and wet lab teams should have, and brainstorming ideas to further strengthen digzyme’s competitive advantages.
— I’m very interested in the ideas for further strengthening digzyme’s competitive advantages.
Absolutely. Regarding how we can grow going forward, I want us to thoroughly discuss and develop the necessary software and wet lab technologies in the business areas that the divisions have identified as targets for expansion.
At the same time, I personally focus on ensuring that the foundational tools and platforms we build within each project are effectively utilized to drive the progress of those projects.
— Nakamura-san, what aspects of working at digzyme do you find most rewarding?
I find it very rewarding to apply new technologies to real-world challenges, developing and updating the missing pieces as we go.
For example, in developing digzyme Spotlight™, we were pioneering the use of AI and machine learning to improve enzymes at a time when almost no one else in the world was doing this.
Before that, it was common to study enzyme structures and conduct research like,
"If we change the part of the protein that interacts with the substrate, the substrate might be affected as well, so let’s mutate it to improve activity."
However, AI and machine learning methods were still not widely adopted.
At the same time, there were many requests to improve enzyme performance—it was a challenge across the entire enzyme industry.
I enjoy facing challenges and solving problems, so I found it very rewarding to create and develop programs while discussing ideas like,
"With digzyme, we should be able to predict and design mutants that increase activity using AI."
But honestly, I enjoy tackling problems of any size, whether big or small.
The Spotlight project was somewhat large scale, but even fixing small, everyday annoyances—things like "this is kind of a hassle"—and watching the system run more smoothly is something I really like.
No matter the scale, I find great satisfaction in new technology development and problem-solving—the process of continuous improvement.
— I see. It sounds like digzyme is really supported by you, Nakamura-san, who finds fulfillment in solving challenges big and small! Since we’re on this topic, I’d love to hear more about the uniqueness of Spotlight.
I’ve heard that the platform was developed by a team that fully leveraged each member’s unique background. Could you please tell us more about this aspect?
Spotlight is a program that uses machine learning algorithms to predict
“If we do it this way, we should be able to identify which parts of the enzyme to modify.”
I had been studying machine learning throughout my student days and also while working at a pharmaceutical company, so I was able to apply that knowledge to create the platform.
Regarding members who are experts in sequence analysis, there’s Mr. Torai and Mr. Hikoyuu⁸ (Informatics Specialist Mr. Hikoyuu Suzuki). They have been studying genome-level gene and protein sequence analysis extensively in the lab.
Then there’s Mr. Tamura⁹ (Informatics Specialist Mr. Koichi Tamura), who is highly knowledgeable about three-dimensional structural data.
So the three of them—Torai, Hikoyuu, and Tamura—worked on determining which features the model should learn from, combining sequence and structural data expertise. Meanwhile, I focused on conceptualizing the machine learning models and approaches.
Finally, Mr. Isozaki implemented the system, and that’s how Spotlight was completed.
— It’s truly moving to hear how you’ve brought together such collective wisdom. May I ask about the challenges you’ve faced at work, and what helped you overcome them?
Rather than challenges per se, I’d say that recruitment has been quite tough.
It’s a significant matter both for the company and for the candidates whose lives are deeply impacted, so I recognize it as a serious responsibility.
In that context, I struggled a lot with how to make the right decisions when hiring people who will shape digzyme’s future.
After several rounds of recruitment, I feel like I’ve finally gotten the hang of it.
Mr. Torai has a very good sense of how to conduct interviews and ask questions, so I’ve learned a lot by following his example.
As for what kind of people we specifically look for, it definitely comes down to those who don’t give off any sense of incongruity during conversations.
We want candidates who not only respond within expected parameters but can also go beyond that in their answers—those are the people we want to hire. On the other hand, if their answers seem stuck one or two steps behind what we expect, it’s a bit difficult to move forward with them.
It’s also important that candidates are good at troubleshooting. Wet lab research especially comes with its share of failures.
In dry lab work, if something goes wrong, you can usually retry quickly—and that’s a field where I tend to come up with ideas easily. But with wet experiments, if you want to redo something, you might lose a whole week, causing significant schedule shifts.
Honestly, I’m not that well-versed in wet lab work myself, so when something goes wrong, it’s important for me to have someone who knows more than I do and can think and act independently during the problem-solving phase.
Troubleshooting experimental issues happens quite often, so we try to hire people who can handle these situations well.
We ask candidates how they’ve handled failures in the past to assess their troubleshooting skills and make sure we bring in capable individuals.
— I see. Since you mentioned WET lab, it makes sense that digzyme’s WET capabilities are so impressive given the team you’ve built.
Exactly. I believe one of digzyme’s strengths in WET lab is that our team can handle a surprisingly wide range of tasks.
For example, when we want to evaluate a certain enzyme, we read relevant papers, develop protocols, try experiments, express proteins, and perform the evaluation. Of course, this requires researchers who can actually conduct solid scientific work.
It’s not something anyone can just casually do by saying, “Hey, read this paper and try replicating the experiment!” (laughs)
Being able to handle that naturally is actually a very high-level skill.
Conversely, it’s very rare that our dry lab analysis gets stalled because of issues on the wet lab side. I say this casually, but it’s actually a remarkable achievement.
That said, while our technical skills are very high, we’re not particularly strong in terms of resources. Compared to many companies and academic labs, we don’t possess special microbial strains or proprietary genetic engineering techniques. We primarily use publicly available materials.
So, honestly, we don’t have an edge in terms of resources, but I take pride in the strong abilities of our research staff.
— I see. That’s reassuring to hear.
Yes. By the way, in DRY lab work, we often don’t know the exact causal relationships. There are many uncertainties about which is the cause and which is the effect.
So when we analyze enzymes, we proceed while considering the possibility of false positives.
We narrow down candidates to very promising ones, but after that, we rely heavily on the high level of WET lab expertise.
For example, even when using E. coli, they don’t just use one strain—they prepare multiple strains, as well as various other organisms—and skillfully conduct experiments to overcome challenges. That expertise is invaluable.

— Looking ahead, what kinds of challenges would you like to take on?
Basically, I hope the projects we’re working on progress through their stages and eventually get launched as products that genuinely make a difference.
I’m excited about the enzymes we’re currently developing becoming actual products — it would be great to say, “This enzyme is actually in that product!” someday.
From a technical standpoint, as projects advance, new challenges unique to those stages will arise, and I want to tackle those.
For example, when you want to convert a certain compound into another, the numerical goals like “the amount of enzyme required” or “the efficiency needed” will become more concrete than they are now.
Achieving—or not achieving—those targets will be a crucial issue in the near future.
The next stage will be “mass production.” We will need to set production performance goals such as “to meet this product price, the culture medium must be this volume,” or “we need to produce this much enzyme.”
Since these goals directly impact business continuity, we must resolve them properly.
Also, I’d love to try developing completely artificial enzymes.
By “completely,” I mean—it’s a tough challenge (laughs). Usually, we base enzyme design on natural enzymes found in microbes or improve upon them. But now, with AI technology, it’s becoming possible to design enzymes from scratch, purely from data.
This means you could design enzymes on a computer that aren’t really based on any natural microbial enzyme, though they might bear some resemblance.
Of course, whether people would want to eat food containing such artificially designed enzymes is another question (laughs).
Currently, we’re bound by natural enzymes as the base, but this approach allows us to break free and create entirely original enzymes.
Even if practical use is uncertain, it’s exciting because it feels truly novel.
I’m also interested in developing systems that don’t rely on microbes, like cell-free systems.
We’re already exploring and discussing cell-free approaches internally, but basically, wet lab processes still mostly involve genetically modifying microbes to express proteins. Often, “protein expression fails,” which is a big hurdle.
Cell-free systems can sometimes reduce that problem—though expression failure can also happen there—so I want to try that.
In any case, I hope to eliminate the unique uncertainties of bio processes.
— When you say "the unique uncertainties of bio processes," what do you mean exactly?
In biological experiments, there are often cases where things just don’t work well, and nobody really knows why.
I think it would be amazing if that “I don’t really understand why it’s failing” part could be eliminated.
I’m not saying we already have a way to guarantee success—this is really just a dream at this point (laughs).
For example, take just the “culture conditions.”
There’s no theoretical way to know exactly which culture conditions are best. When culturing a microbe, you repeatedly try different combinations of ingredients in the medium and experimentally find which one works best.
It’s not like you can say, “This is the best condition” based purely on theory—it’s more like, “I don’t really know why, but this works better.”
Sometimes the culture grows well, and sometimes it doesn’t. Protein production is similar—it’s not consistent; sometimes you get a lot, other times less. There’s quite a bit of variation.
Biological experiments inherently have these fluctuations.
“Failure” is an extreme example of this uncertainty, but even when things are going well, there are times when it’s “especially good” and times when it’s “just okay,” so there’s a lot of variability and error.
Even amid these uncertainties, one major challenge remains “poor expression depending on the host organism.”
To improve this, we often think, “It would be great if there was a system that could express any enzyme from any organism.”
If such a system exists, I feel that cell-free systems might be the answer.
— Expression of anything—that’s quite a dream. Thank you for the detailed explanation. Lastly, do you have a message for future team members considering applying to digzyme?
“Let’s tackle the ambiguous challenges of biology together with cutting-edge technology and innovative ideas!”
— Thank you very much, Mr. Nakamura.
¹ Nao Torai – CEO and co-founder of digzyme Inc.
² Dr. Takuji Yamada – Associate Professor at Tokyo Institute of Technology and CSO of digzyme Inc.
³ Yamada Lab – Laboratory for Life Science and Technology at Tokyo Institute of Technology.
⁴ NEDO: Japan’s national agency for promoting research and development of new energy and industrial technologies.
⁵ spotlight: digzyme’s platform for enzyme function improvement through machine learning.
⁶ Principal Investigator Mr. Tatsuhiro Isozaki
⁷ Principal Investigator Mr. Yuki Takayama
⁸ Informatics Specialist Mr. Hikoyuu Suzuki
⁹ Informatics Specialist Mr. Koichi Tamura
Closing Remarks
▼ Original article is available here (note)
https://note.com/digzyme/n/n4cb24197110b
Expected Practical Applications of the digzyme Custom Enzyme Lab: Approaches to Glycan Structure Construction and Recalcitrant Substance Degradation
Introduction
From May 21 (Wed) to May 23 (Fri), 2025, ifia JAPAN 2025 was held over three days.
As with last year, our CEO, Dr. Watarai, gave an exhibitor presentation at the event.
The full presentation is now available on YouTube—please feel free to take a look.
In this exhibitor presentation, we introduced the newly launched “digzyme Custom Enzyme Lab,” unveiled on May 21, 2025.
The session covered two key technological approaches: DRY (bioinformatics-based analysis) and WET (experimental validation), and provided an overview of the entire platform.
This article takes a deeper dive into two potential real-world applications of the digzyme Custom Enzyme Lab, which were briefly mentioned during the presentation.
Through a Q&A format and from the perspective of our CEO Dr. Watarai, we explore the technical breakthroughs behind each case, as well as the in silico design strategies employed.
While the presentation offered a high-level overview, this article aims to give you a more concrete understanding of the capabilities and potential of the digzyme Custom Enzyme Lab.
We invite you to read on and explore the details—beginning with the first case study.

Expected Application Case 1 of the digzyme Custom Enzyme Lab
Q: What do you consider the most significant value of this result?
A: The physical properties of carbohydrates vary depending on the linkage patterns between constituent monosaccharides.
This case is particularly valuable because it represents a rare example—even in academic contexts—where in silico techniques successfully identified an enzyme capable of constructing a specific glycan structure.
Moreover, the target enzyme was discovered with just 10 experimental validations, which highlights the efficiency and precision of the approach.
Q: What was innovative about this approach compared to conventional methods?
A:In this case, our proprietary, detailed analytical techniques ultimately proved effective when applied to the deep learning (DL)-based structural prediction technologies of the time, such as AlphaFold2. Traditional homology-based models had difficulty predicting subtle structural differences in proteins that lead to variations in glycan structures. However, the AI technologies available at the time enabled us to capture some of these critical features to a certain extent.
(Note: As there is still a gap between these earlier AI technologies and today's cutting-edge generative models, we use the term "AI" here for convenience.)
Q: What team efforts or contributions led to this success?
A: The lead researcher deeply investigated the client’s specific needs and successfully translated them into tailored screening criteria for enzyme selection.
By working closely with our core development team, a customized analysis pipeline was developed, which was crucial to achieving this outcome.
We believe one of our key strengths is the ability to flexibly build new tools and solutions beyond our existing platforms to meet unique and complex challenges.
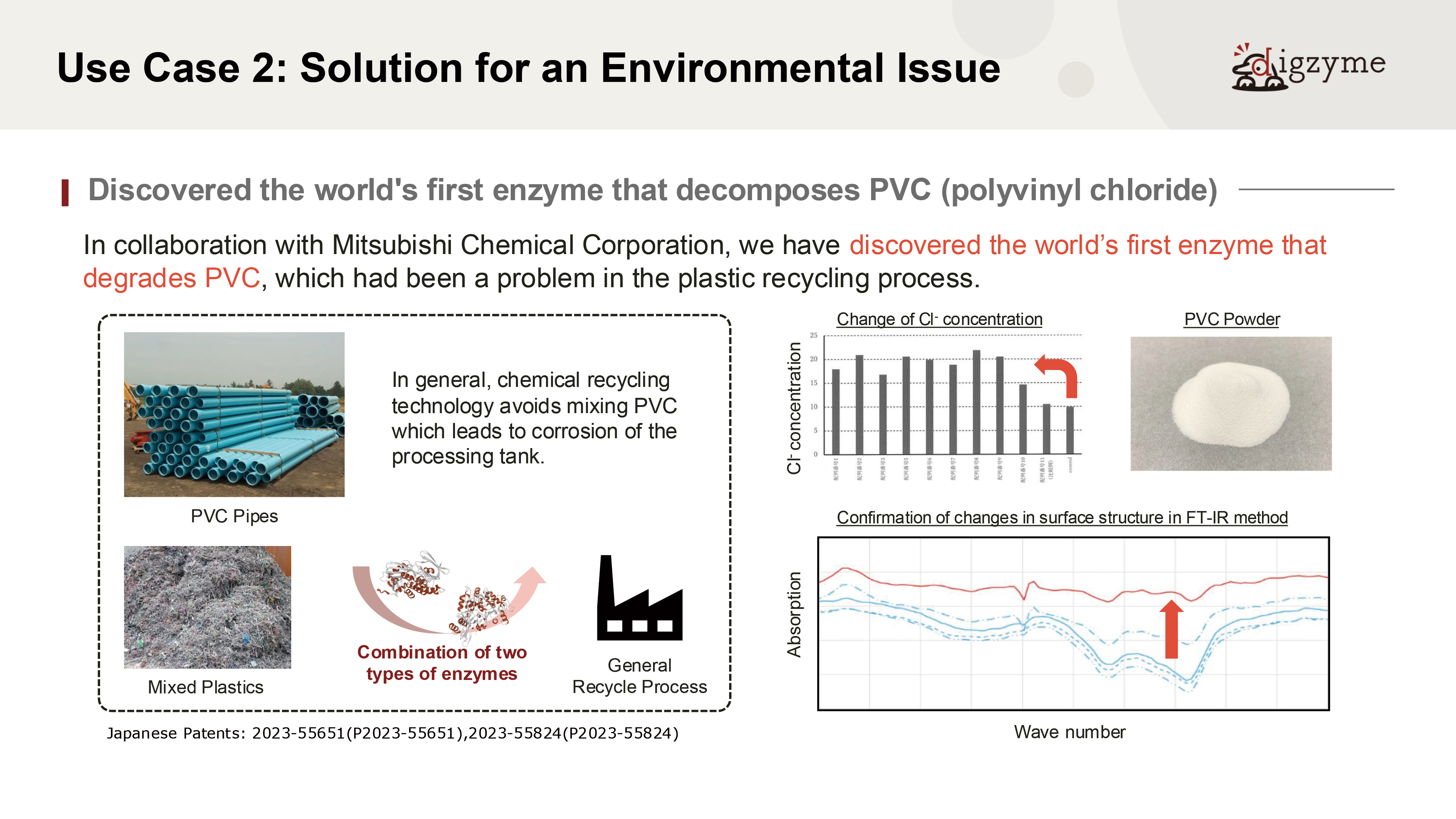
Next, let us introduce the second case study, which was conducted in collaboration with Mitsubishi Chemical Corporation.
Expected Application Case 2 of the digzyme Custom Enzyme Lab
Q: What do you consider the most significant value of this result?
A: PVC (polyvinyl chloride) is a synthetic compound whose mass production began in the 20th century and does not exist in nature.
Assuming that natural microorganisms have not evolved degradation mechanisms for such materials, it would be highly unlikely to discover well-optimized degrading enzymes from natural sources.
However, living organisms are known to retain a wide variety of “non-optimized” or dormant genes within their genomes, which may later contribute to adaptation under environmental pressure.
This case can be seen as an attempt to identify such latent enzymatic functions through in silico screening—making it a particularly challenging theme.
Q: How long would it have taken to discover such an enzyme using conventional methods?
A: In recent years, there have been several studies that identify artificial plastic-degrading enzymes using methods akin to enrichment culturing. For example, researchers may submerge a particular type of plastic resin in the seabed for an extended period, then retrieve and observe its degradation, or isolate and culture microbes from biofilms formed on the plastic.
When successful, these efforts can uncover microorganisms with plastic-degrading enzymes, allowing identification through genomic analysis or BAC library construction. However, due to the inherently slow degradation process, such approaches often require years to yield results.
Moreover, it is common for degradation not to occur at all, resulting in unsuccessful attempts. In contrast, in silico discovery can typically be completed within about six months, making it a relatively efficient method even for targets that would otherwise require long-term experimental work.
Conclusion
Reflecting on the presentation, Dr. Watarai shared the following comment:
“With digzyme Custom Enzyme Lab, we are able to prepare in silico libraries in advance—similar to what we did in these collaborative cases. It’s a service we recommend to customers seeking to test purified enzymes from high-precision candidate libraries.”
As this statement illustrates, a bioinformatics-based approach to enzyme design has the potential to dramatically accelerate practical enzyme development, even under resource-constrained conditions.
As applications continue to expand across diverse domains, digzyme Custom Enzyme Lab is expected to play a pivotal role as a core technological foundation.